

MIRA: joint regulatory modeling of multimodal expression and chromatin accessibility in single cells
- PMID: 36068320
- PMCID: PMC9517733
- DOI: 10.1038/s41592-022-01595-z
MIRA: joint regulatory modeling of multimodal expression and chromatin accessibility in single cells
Abstract
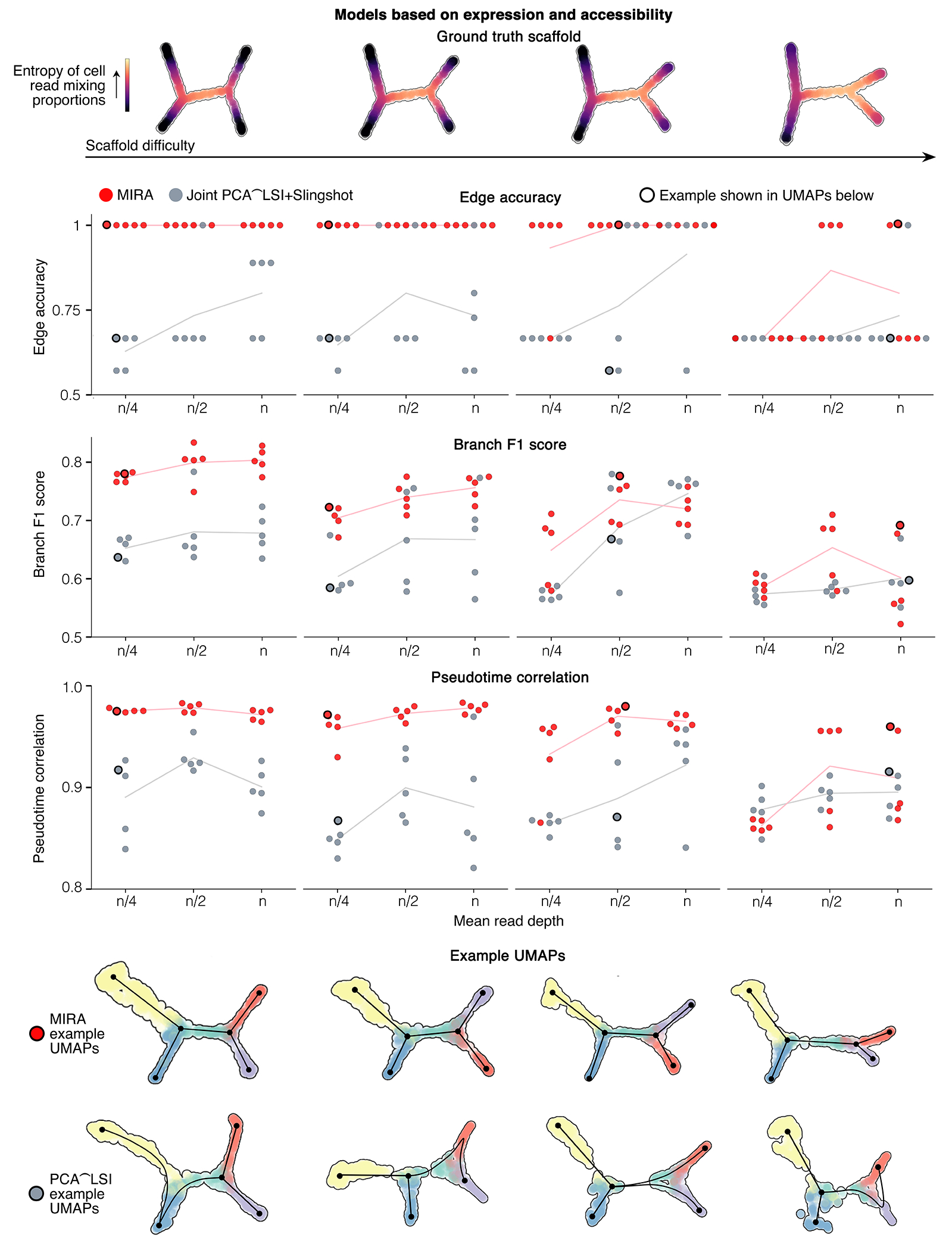
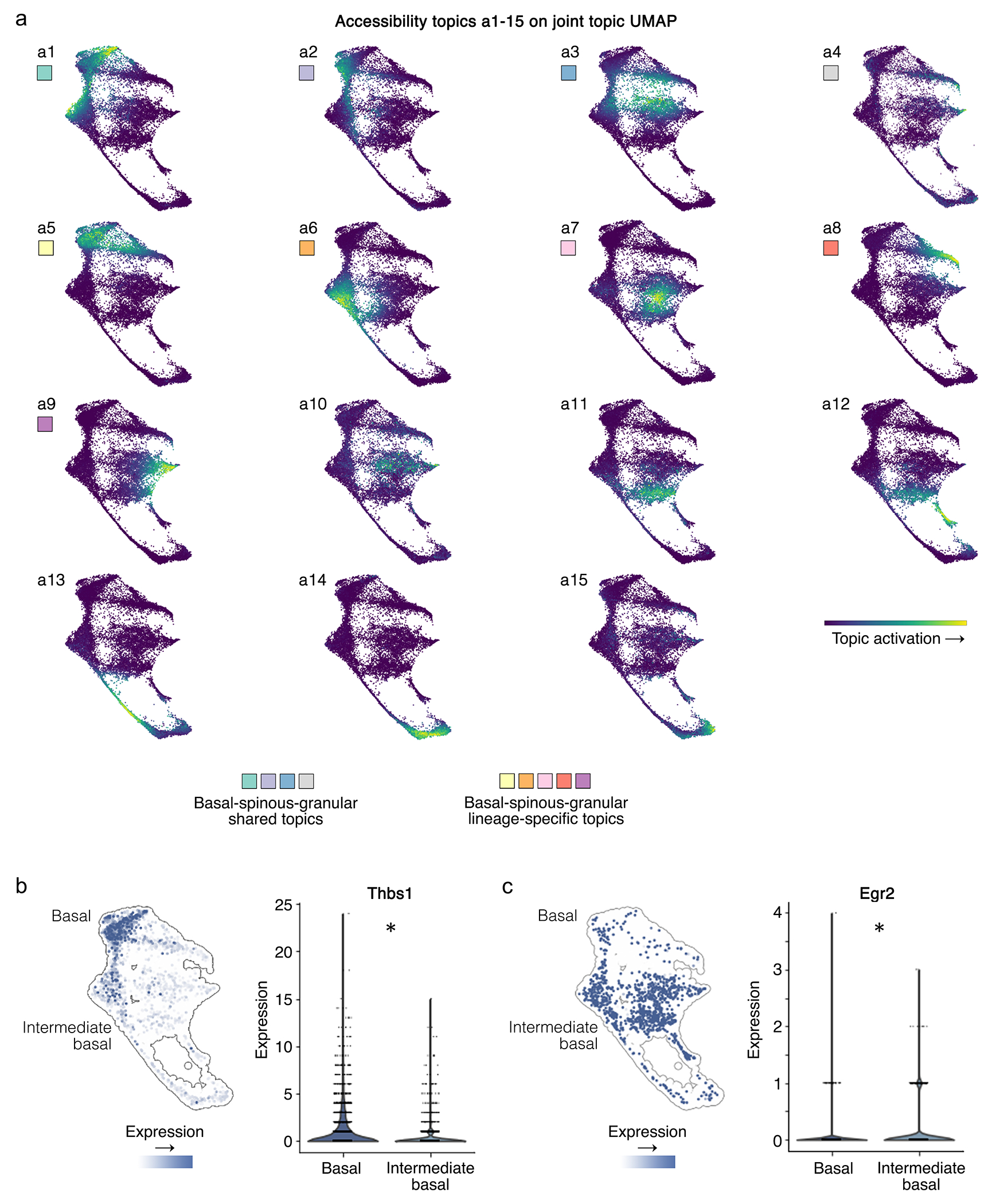
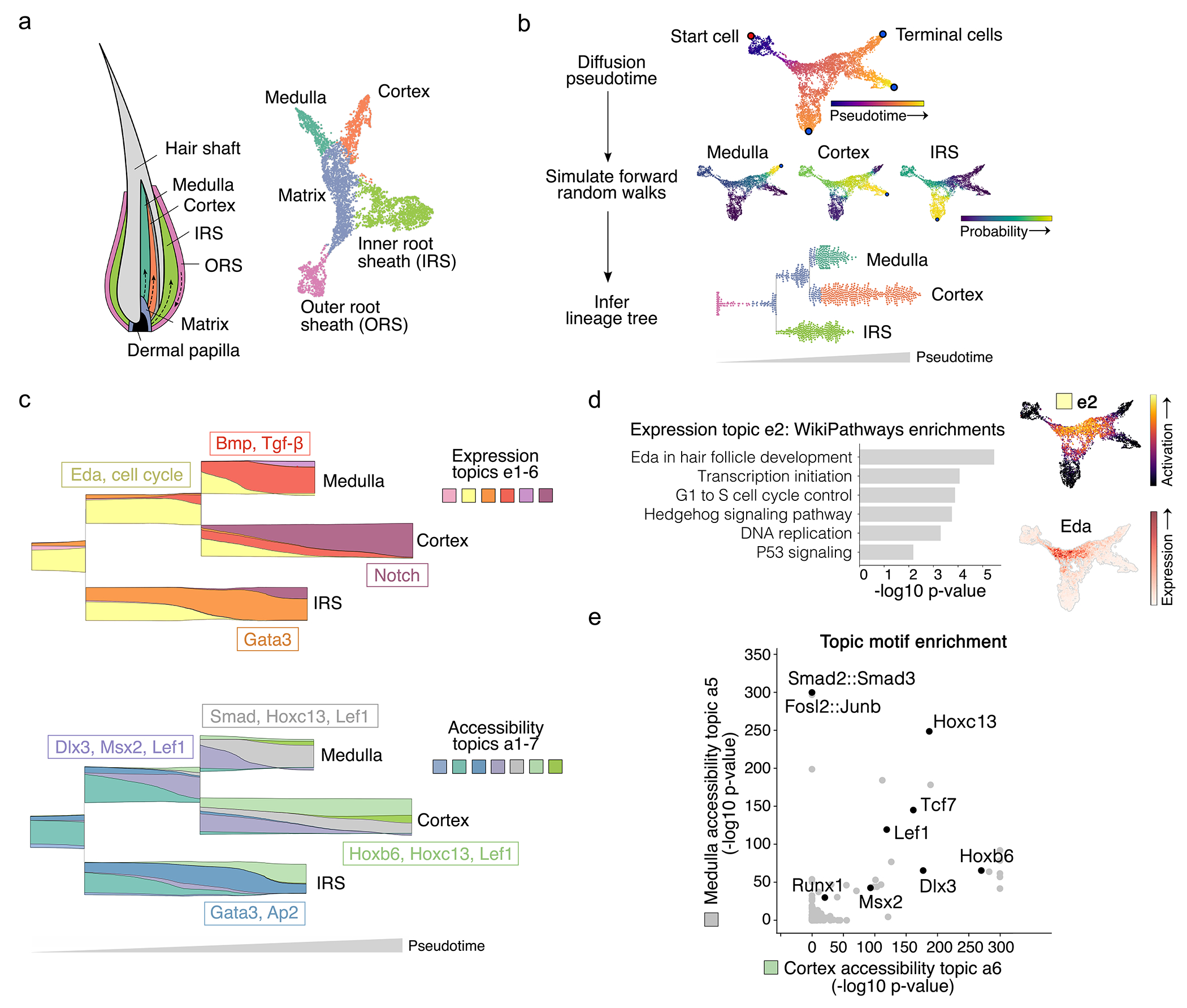
Rigorously comparing gene expression and chromatin accessibility in the same single cells could illuminate the logic of how coupling or decoupling of these mechanisms regulates fate commitment. Here we present MIRA, probabilistic multimodal models for integrated regulatory analysis, a comprehensive methodology that systematically contrasts transcription and accessibility to infer the regulatory circuitry driving cells along cell state trajectories. MIRA leverages topic modeling of cell states and regulatory potential modeling of individual gene loci. MIRA thereby represents cell states in an efficient and interpretable latent space, infers high-fidelity cell state trees, determines key regulators of fate decisions at branch points and exposes the variable influence of local accessibility on transcription at distinct loci. Applied to epidermal differentiation and embryonic brain development from two different multimodal platforms, MIRA revealed that early developmental genes were tightly regulated by local chromatin landscape whereas terminal fate genes were titrated without requiring extensive chromatin remodeling.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
The remaining authors declare no competing interests.
Figures













References
Methods References
-
- Blei DM., Ng AY. & Edu JB. Latent Dirichlet Allocation Michael I. Jordan. Journal of Machine Learning Research 3, 993–1022 (2003).
-
- Kingma DP & Welling M Auto-Encoding Variational Bayes. arXiv:1312.6114 (2013).
