

Treatment of Sindbis Virus-Infected Neurons with Antibody to E2 Alters Synthesis of Complete and nsP1-Expressing Defective Viral RNAs
- PMID: 36069441
- PMCID: PMC9600605
- DOI: 10.1128/mbio.02221-22
Treatment of Sindbis Virus-Infected Neurons with Antibody to E2 Alters Synthesis of Complete and nsP1-Expressing Defective Viral RNAs
Abstract
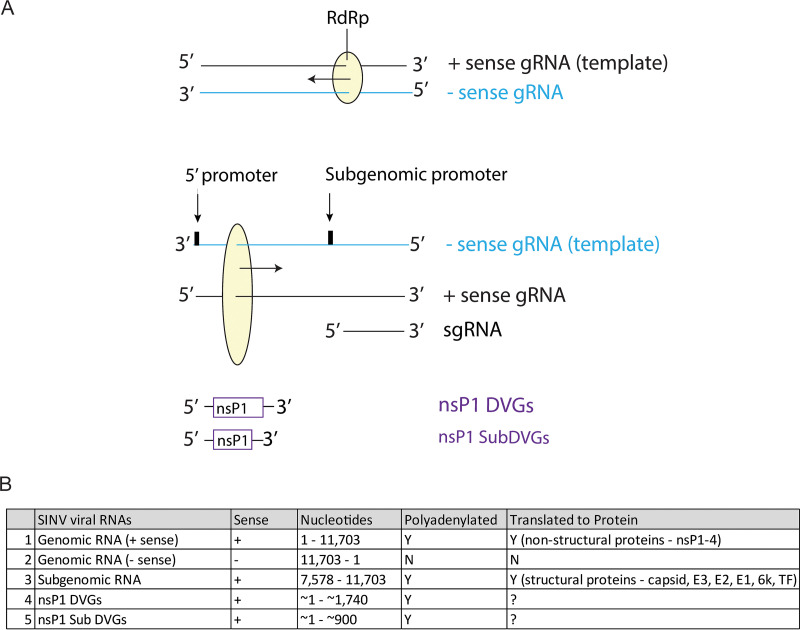
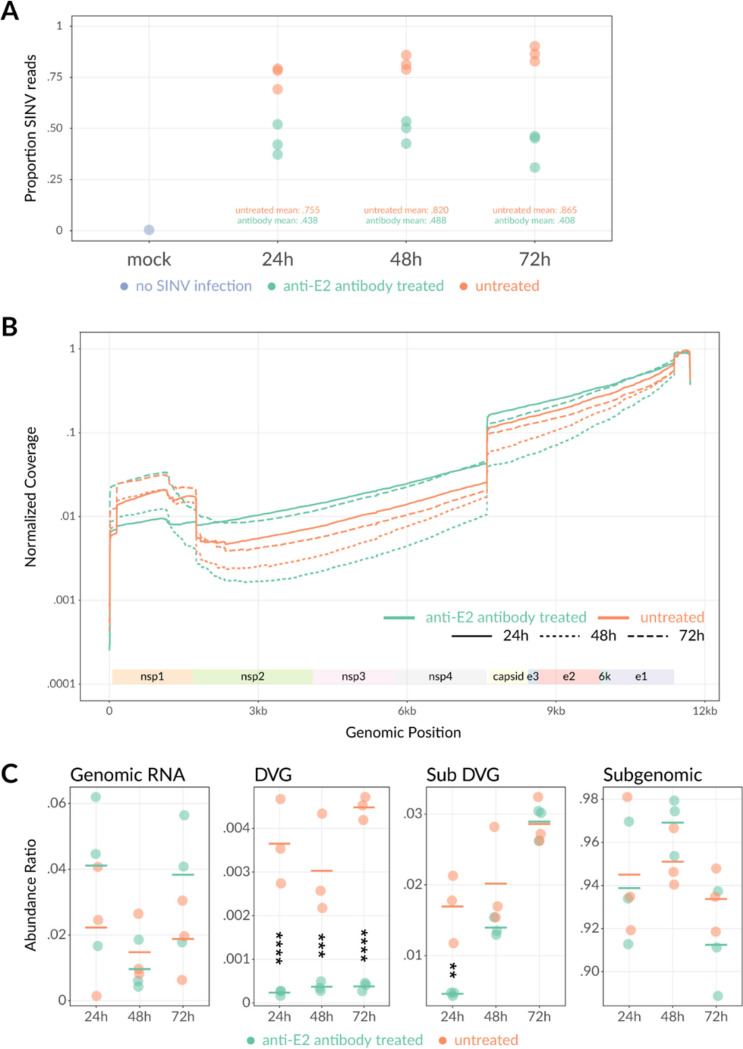
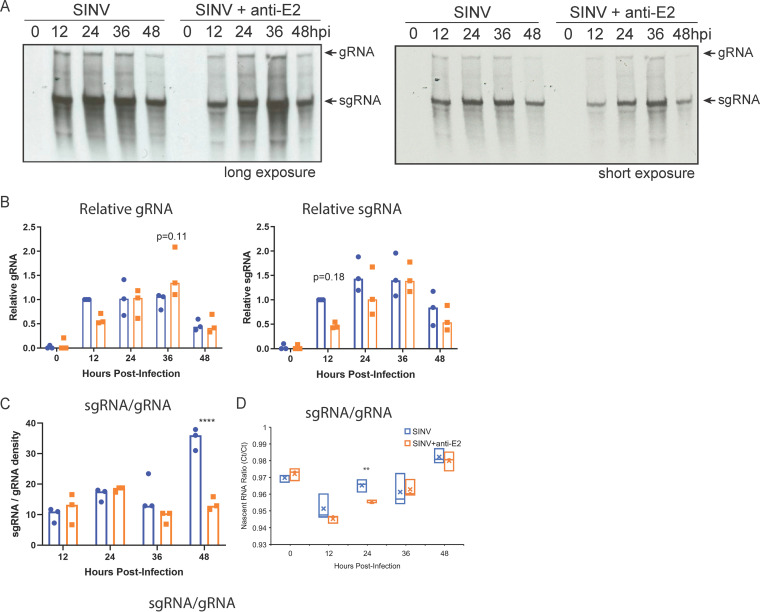
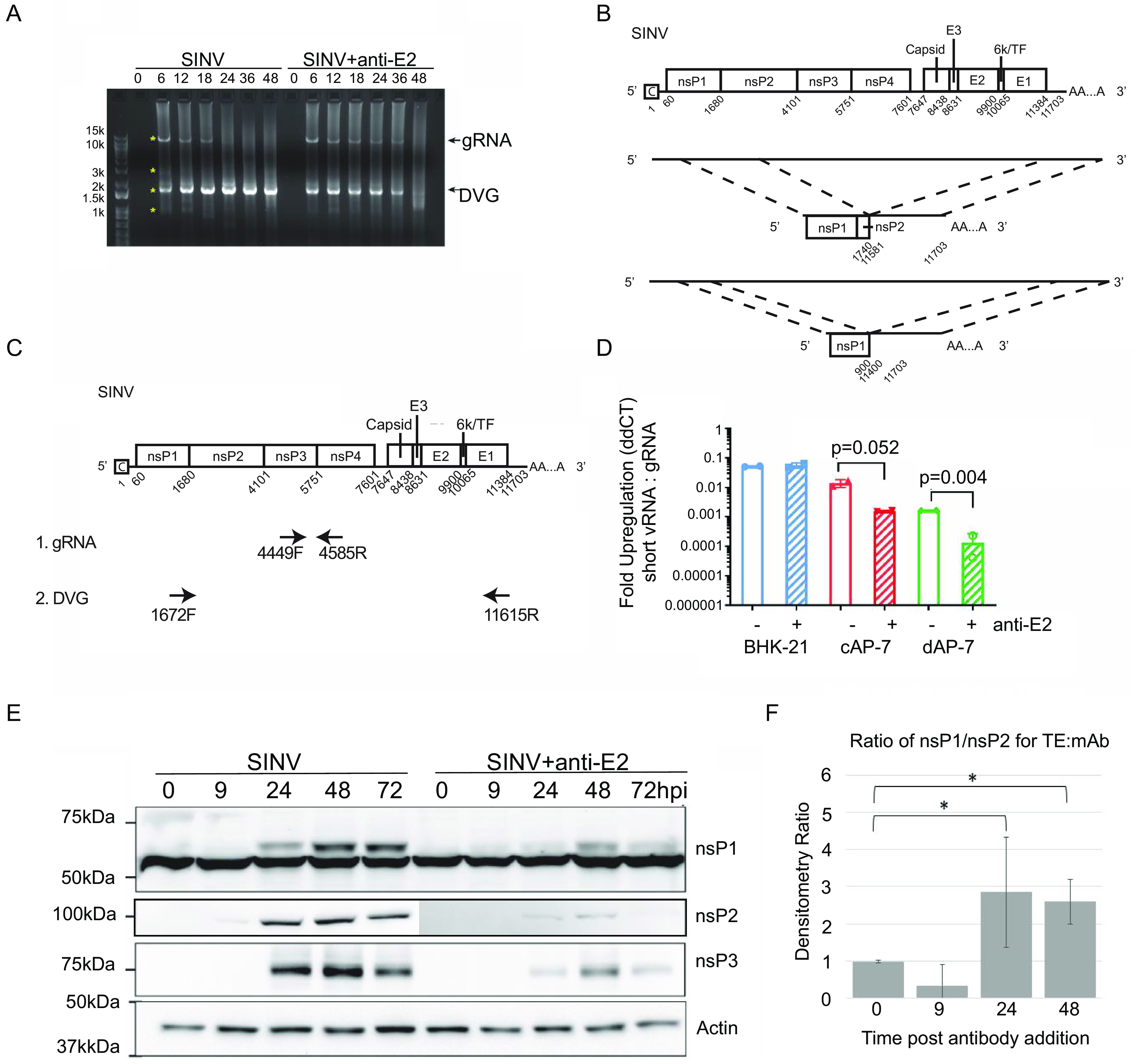
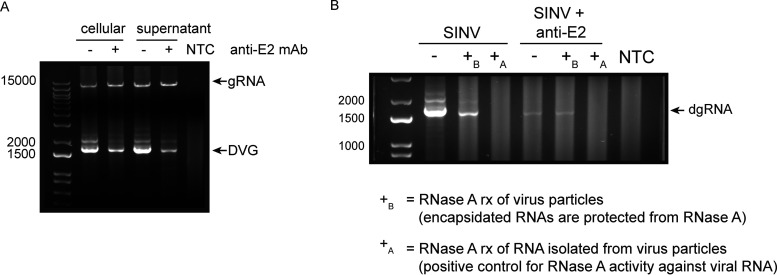
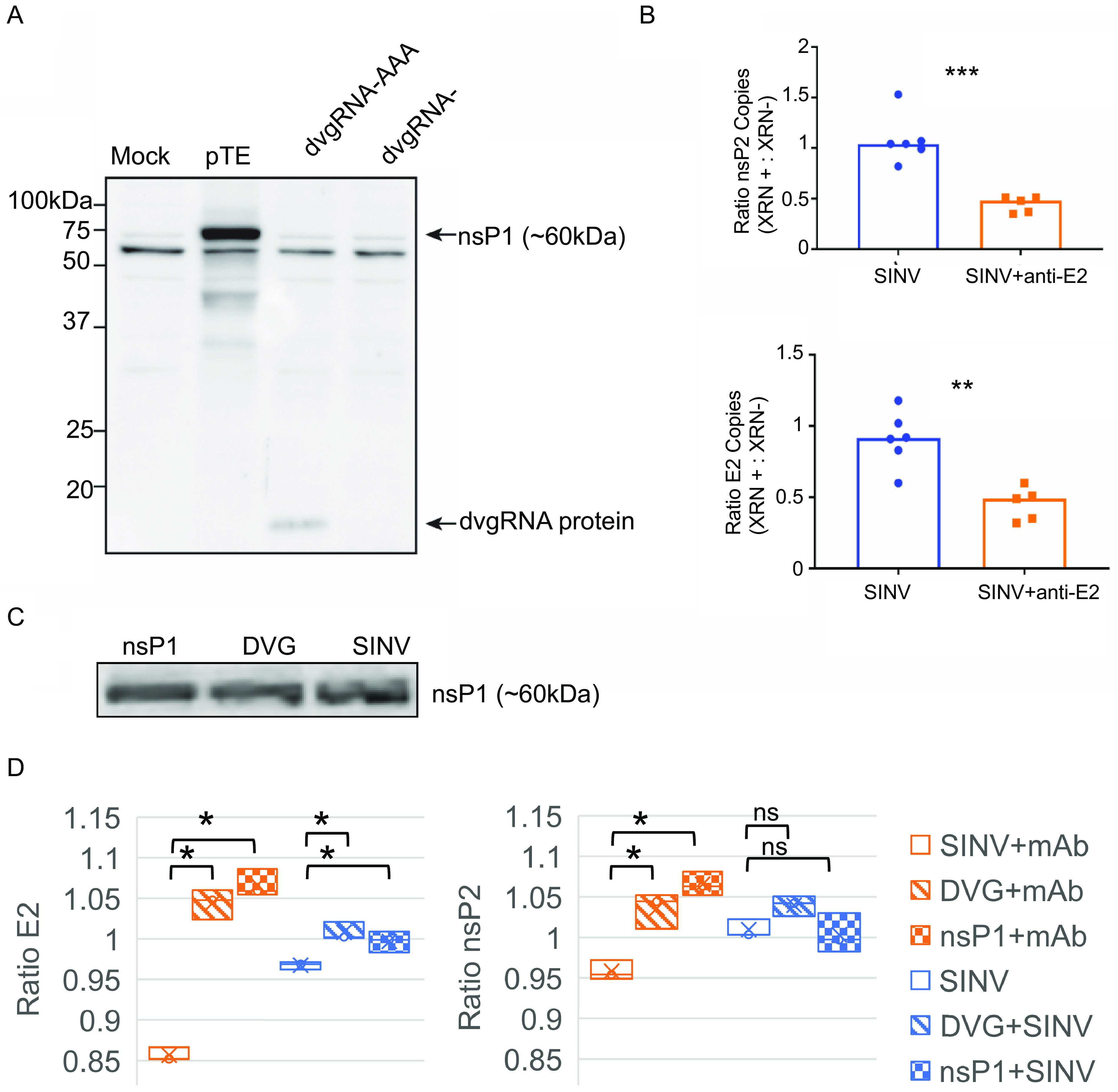
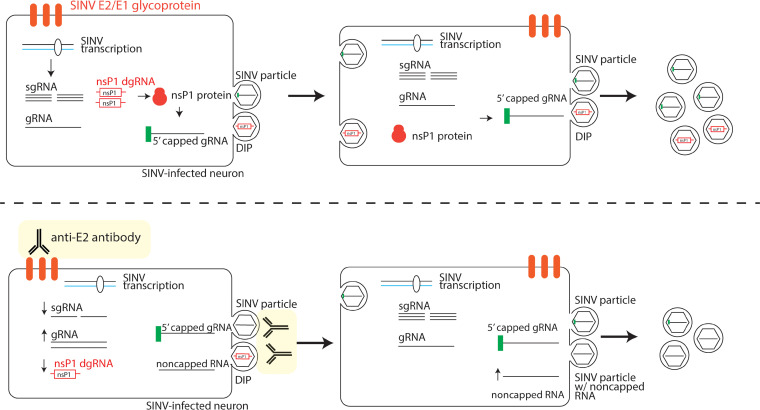
Alphaviruses are positive-sense RNA viruses that are important causes of viral encephalomyelitis. Sindbis virus (SINV), the prototype alphavirus, preferentially infects neurons in mice and is a model system for studying mechanisms of viral clearance from the nervous system. Antibody specific to the SINV E2 glycoprotein plays an important role in SINV clearance, and this effect is reproduced in cultures of infected mature neurons. To determine how anti-E2 antibody affects SINV RNA synthesis, Oxford Nanopore Technologies direct long-read RNA sequencing was used to sequence viral RNAs following antibody treatment of infected neurons. Differentiated AP-7 rat olfactory neuronal cells, an in vitro model for mature neurons, were infected with SINV and treated with anti-E2 antibody. Whole-cell RNA lysates were collected for sequencing of poly(A)-selected RNA 24, 48, and 72 h after infection. Three primary species of viral RNA were produced: genomic, subgenomic, and defective viral genomes (DVGs) encoding the RNA capping protein nsP1. Antibody treatment resulted in overall lower production of SINV RNA, decreased synthesis of subgenomic RNA relative to genomic RNA, and suppressed production of the nsP1 DVG. The nsP1 DVG was packaged into virus particles and could be translated. Because antibody-treated cells released a higher proportion of virions with noncapped genomes and transient transfection to express the nsP1 DVG improved viral RNA capping in antibody-treated cells, we postulate that one mechanism by which antibody inhibits SINV replication in neurons is to suppress DVG synthesis and thus decrease production of infectious virions containing capped genomes. IMPORTANCE Alphaviruses are important causes of viral encephalomyelitis without approved treatments or vaccines. Antibody to the Sindbis virus (SINV) E2 glycoprotein is required for immune-mediated noncytolytic virus clearance from neurons. We used direct RNA nanopore sequencing to evaluate how anti-E2 antibody affects SINV replication at the RNA level. Antibody altered the viral RNAs produced by decreasing the proportion of subgenomic relative to genomic RNA and suppressing production of a previously unrecognized defective viral genome (DVG) encoding nsP1, the viral RNA capping enzyme. Antibody-treated neurons released a lower proportion of SINV particles with capped genomes necessary for translation and infection. Decreased nsP1 DVG production in antibody-treated neurons led to lower expression of nsP1 protein, decreased genome capping efficiency, and release of fewer infectious virus particles. Capping was increased with exogenous expression of the nsP1 DVG. These studies identify a novel alphavirus DVG function and new mechanism for antibody-mediated control of virus replication.
Keywords: alphavirus; defective viral genomes; nanopore sequencing; neuron infection; viral encephalitis; virus clearance.
Conflict of interest statement
The authors declare a conflict of interest. D.E.G. is a member of advisory boards for GlaxoSmithKline, GreenLight Bioscience, Takeda Pharmaceuticals and Academia Sinica. J.X.Y. is an employee of Takeda Pharmaceuticals.
Figures
References
-
- Jackson AC, Moench TR, Trapp BD, Griffin DE. 1988. Basis of neurovirulence in Sindbis virus encephalomyelitis of mice. Lab Invest 58:503–509. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
