

Oncohistone interactome profiling uncovers contrasting oncogenic mechanisms and identifies potential therapeutic targets in high grade glioma
- PMID: 36070144
- PMCID: PMC9547787
- DOI: 10.1007/s00401-022-02489-2
Oncohistone interactome profiling uncovers contrasting oncogenic mechanisms and identifies potential therapeutic targets in high grade glioma
Abstract
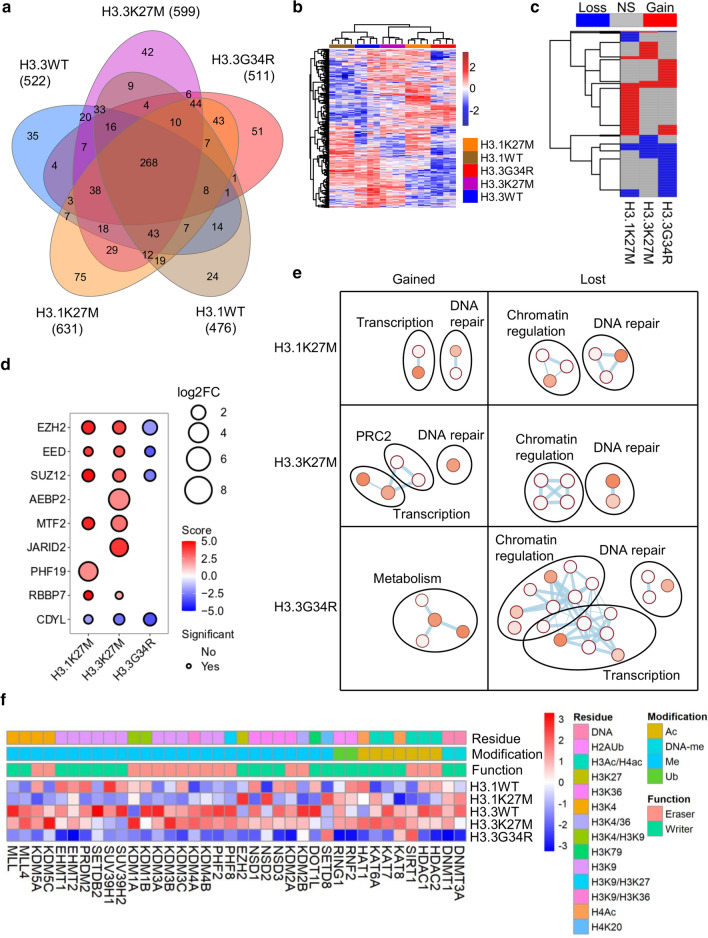
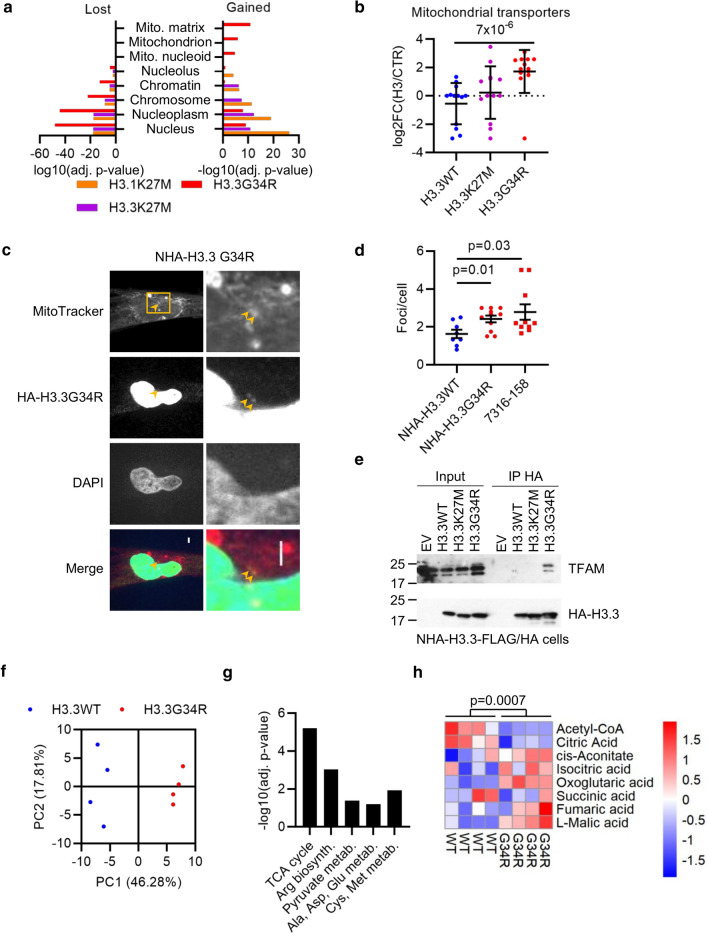
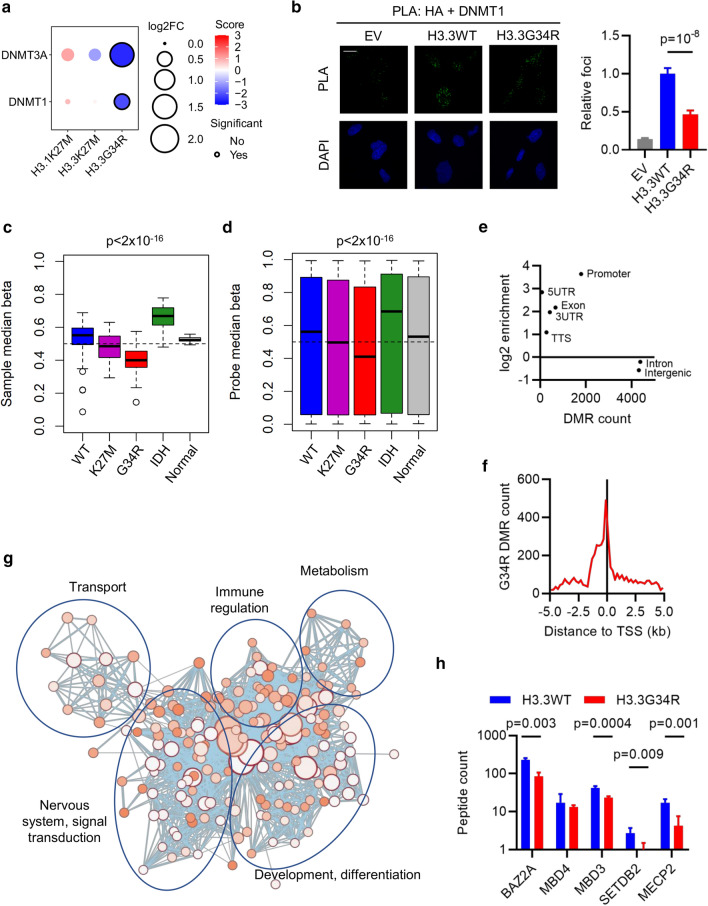
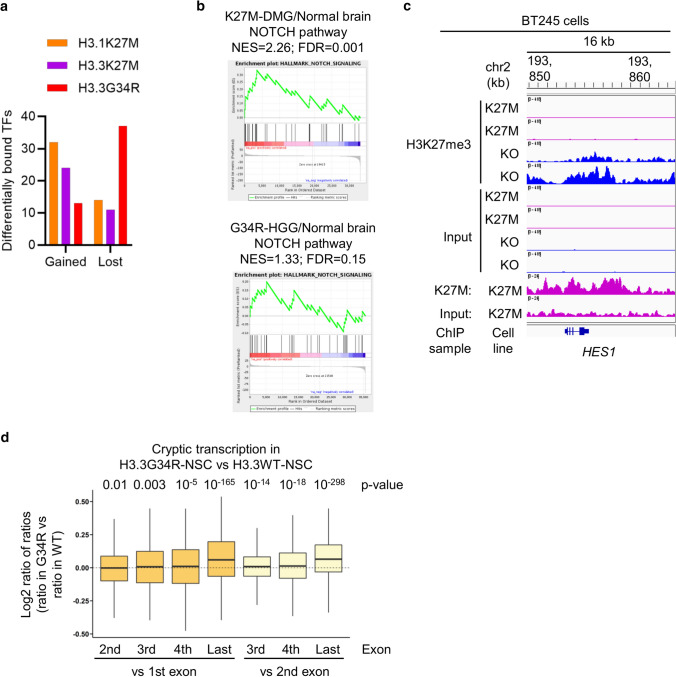
Histone H3 mutations at amino acids 27 (H3K27M) and 34 (H3G34R) are recurrent drivers of pediatric-type high-grade glioma (pHGG). H3K27M mutations lead to global disruption of H3K27me3 through dominant negative PRC2 inhibition, while H3G34R mutations lead to local losses of H3K36me3 through inhibition of SETD2. However, their broader oncogenic mechanisms remain unclear. We characterized the H3.1K27M, H3.3K27M and H3.3G34R interactomes, finding that H3K27M is associated with epigenetic and transcription factor changes; in contrast H3G34R removes a break on cryptic transcription, limits DNA methyltransferase access, and alters mitochondrial metabolism. All 3 mutants had altered interactions with DNA repair proteins and H3K9 methyltransferases. H3K9me3 was reduced in H3K27M-containing nucleosomes, and cis-H3K9 methylation was required for H3K27M to exert its effect on global H3K27me3. H3K9 methyltransferase inhibition was lethal to H3.1K27M, H3.3K27M and H3.3G34R pHGG cells, underscoring the importance of H3K9 methylation for oncohistone-mutant gliomas and suggesting it as an attractive therapeutic target.
Keywords: H3G34R; H3K27M; H3K9 methylation; pHGG.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures
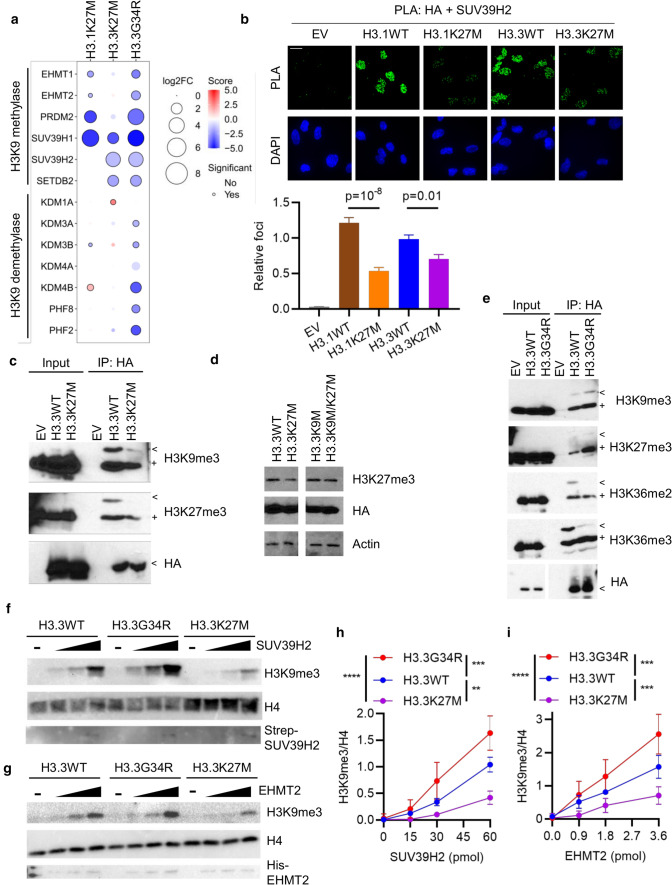
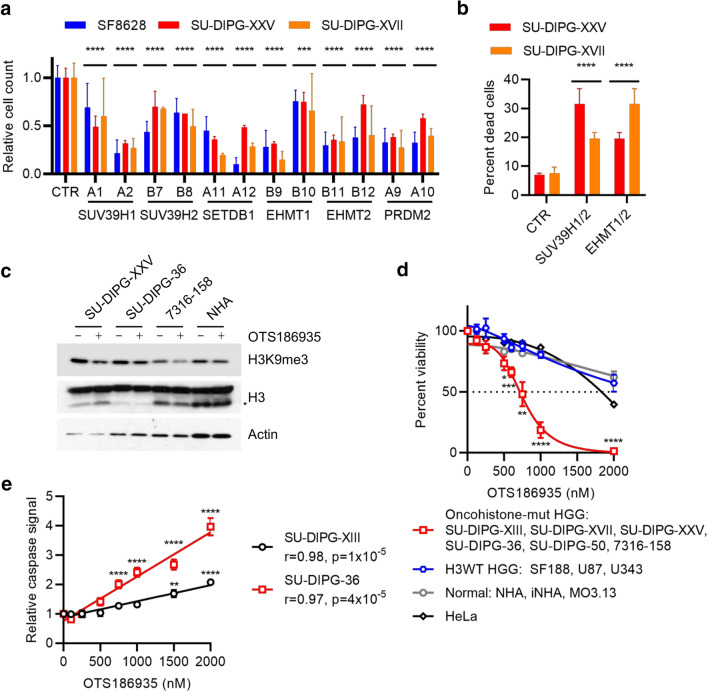
References
-
- Atak ZK, Gianfelici V, Hulselmans G, De Keersmaecker K, Devasia AG, Geerdens E, Mentens N, Chiaretti S, Durinck K, Uyttebroeck A, et al. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet. 2013;9:e1003997. doi: 10.1371/journal.pgen.1003997. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
