

3D chromatin maps of the human pancreas reveal lineage-specific regulatory architecture of T2D risk
- PMID: 36070683
- PMCID: PMC9664375
- DOI: 10.1016/j.cmet.2022.08.014
3D chromatin maps of the human pancreas reveal lineage-specific regulatory architecture of T2D risk
Abstract
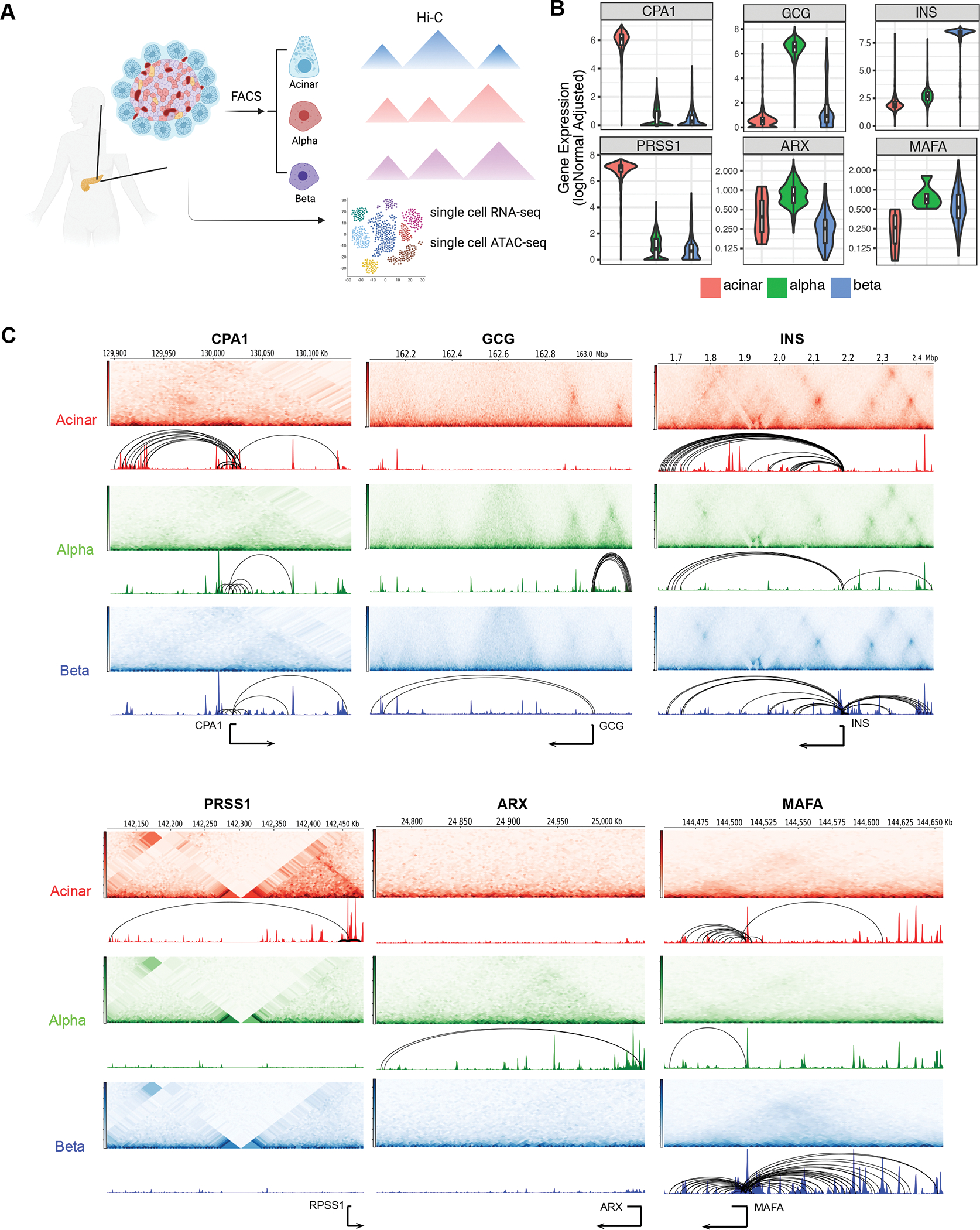
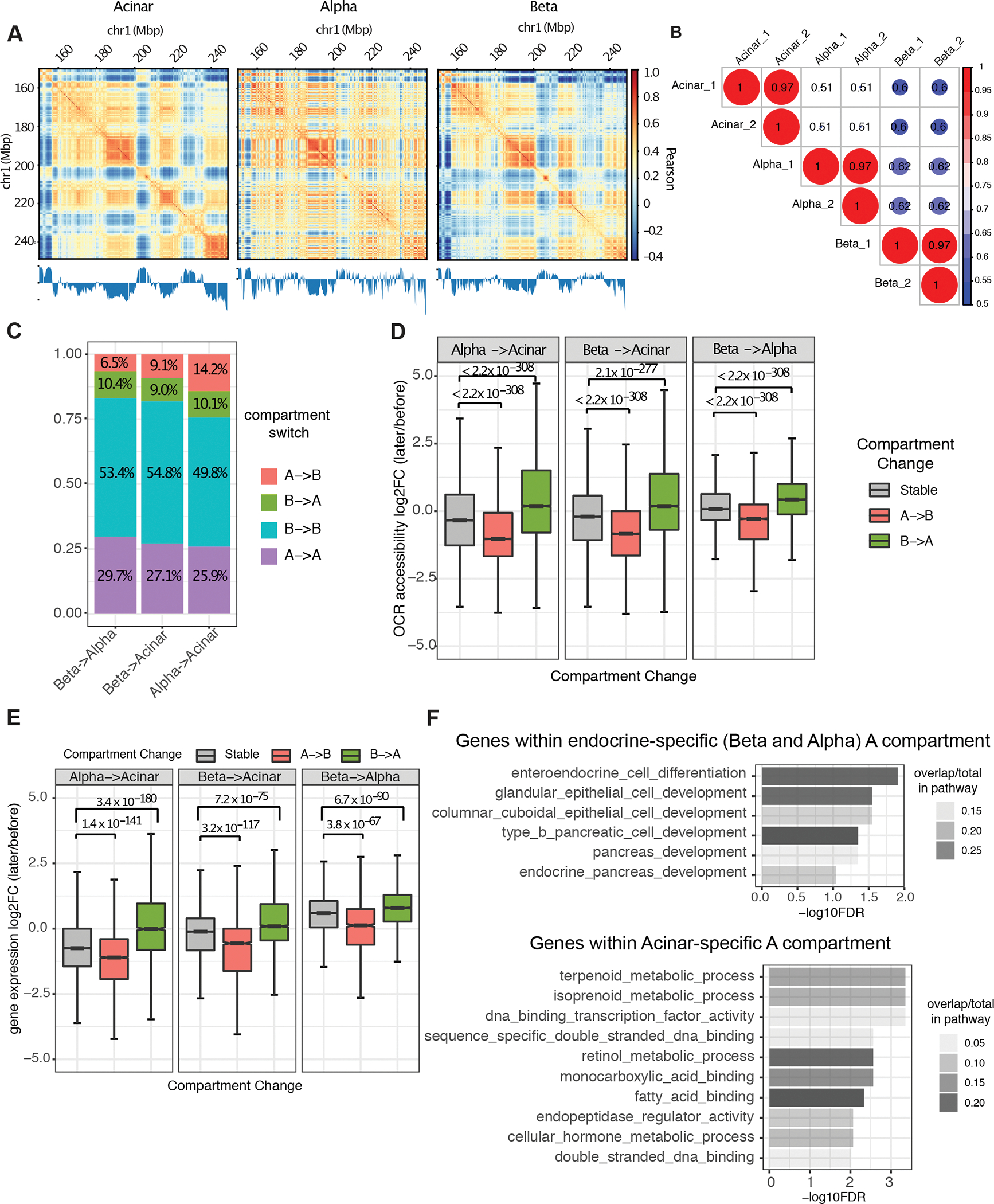
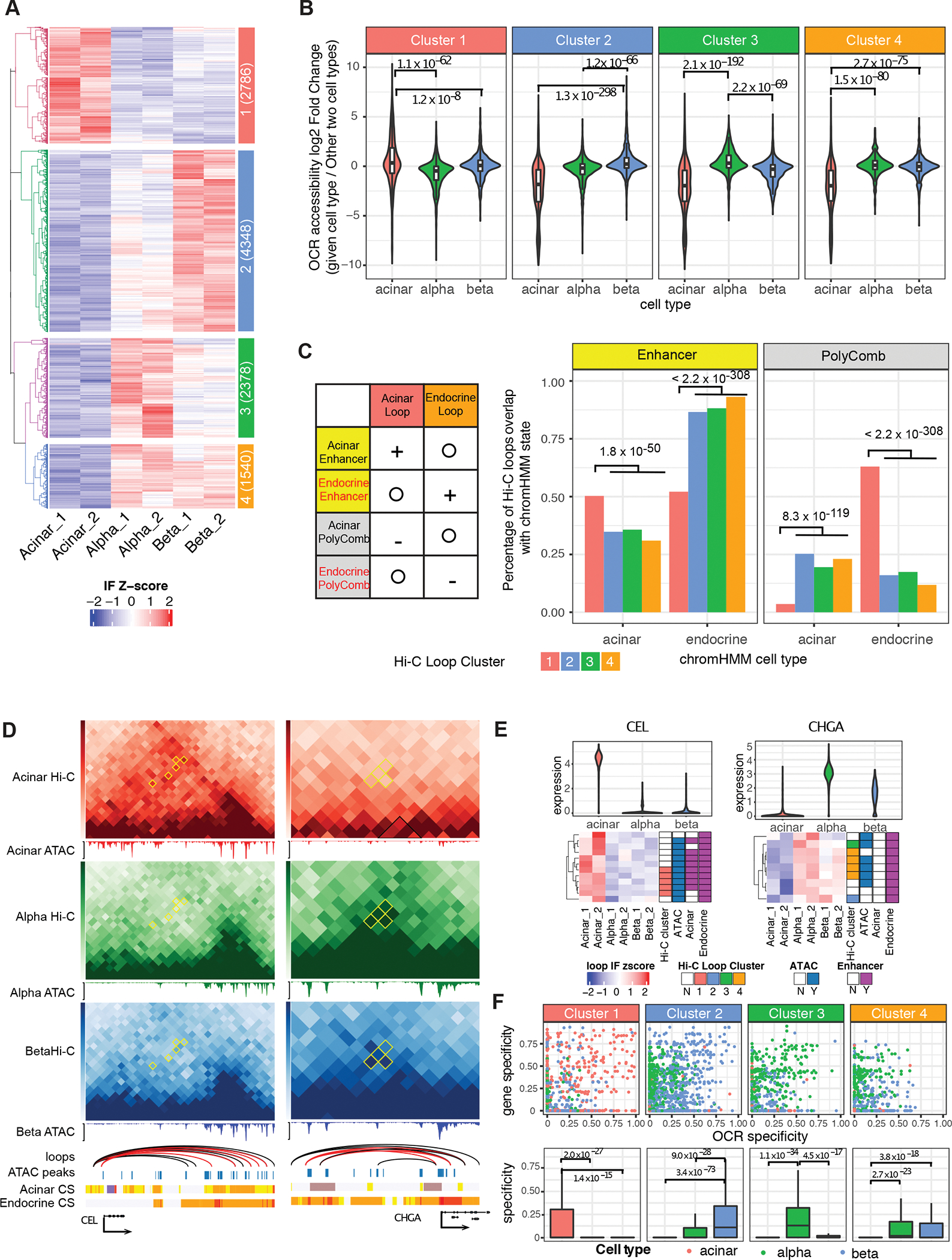
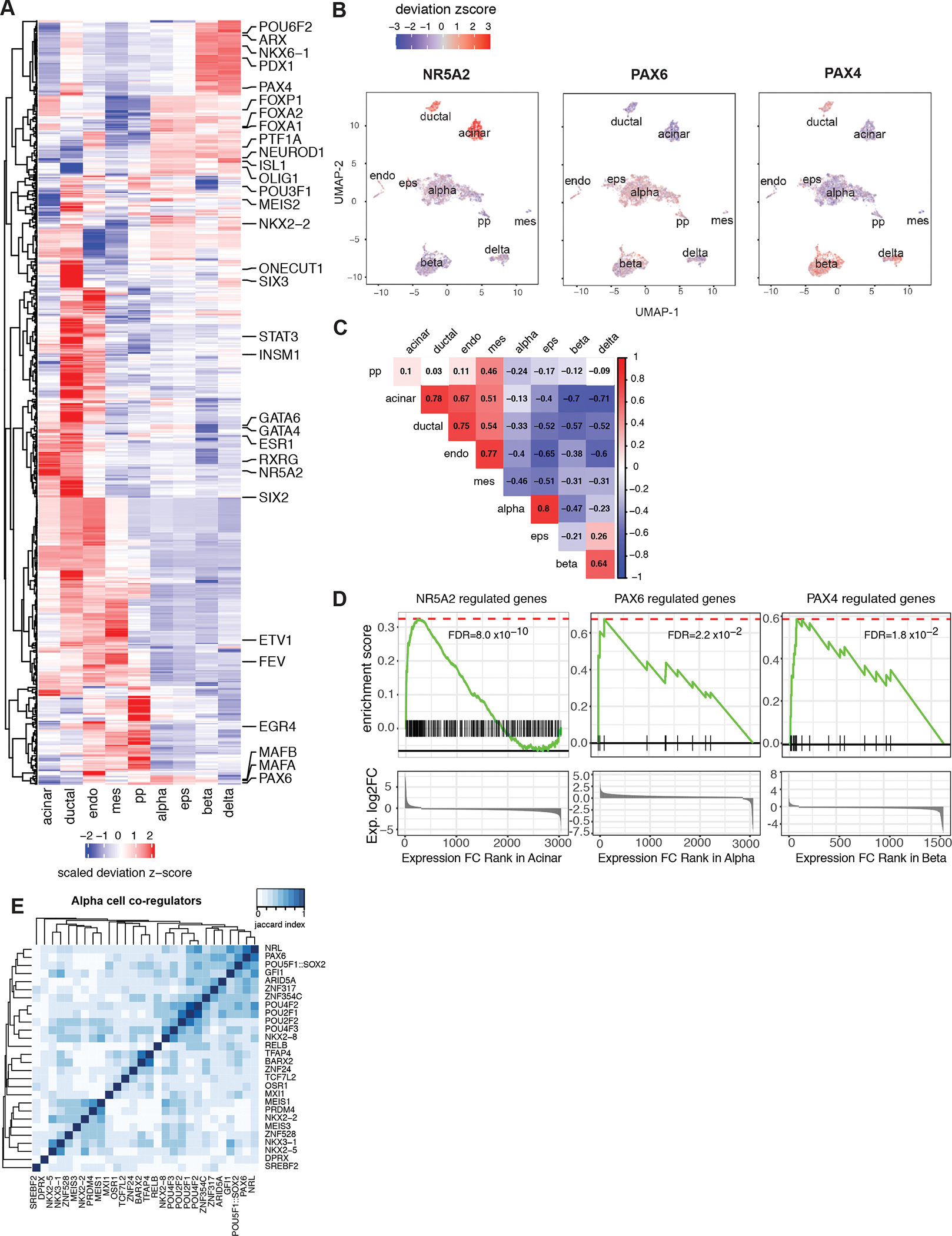
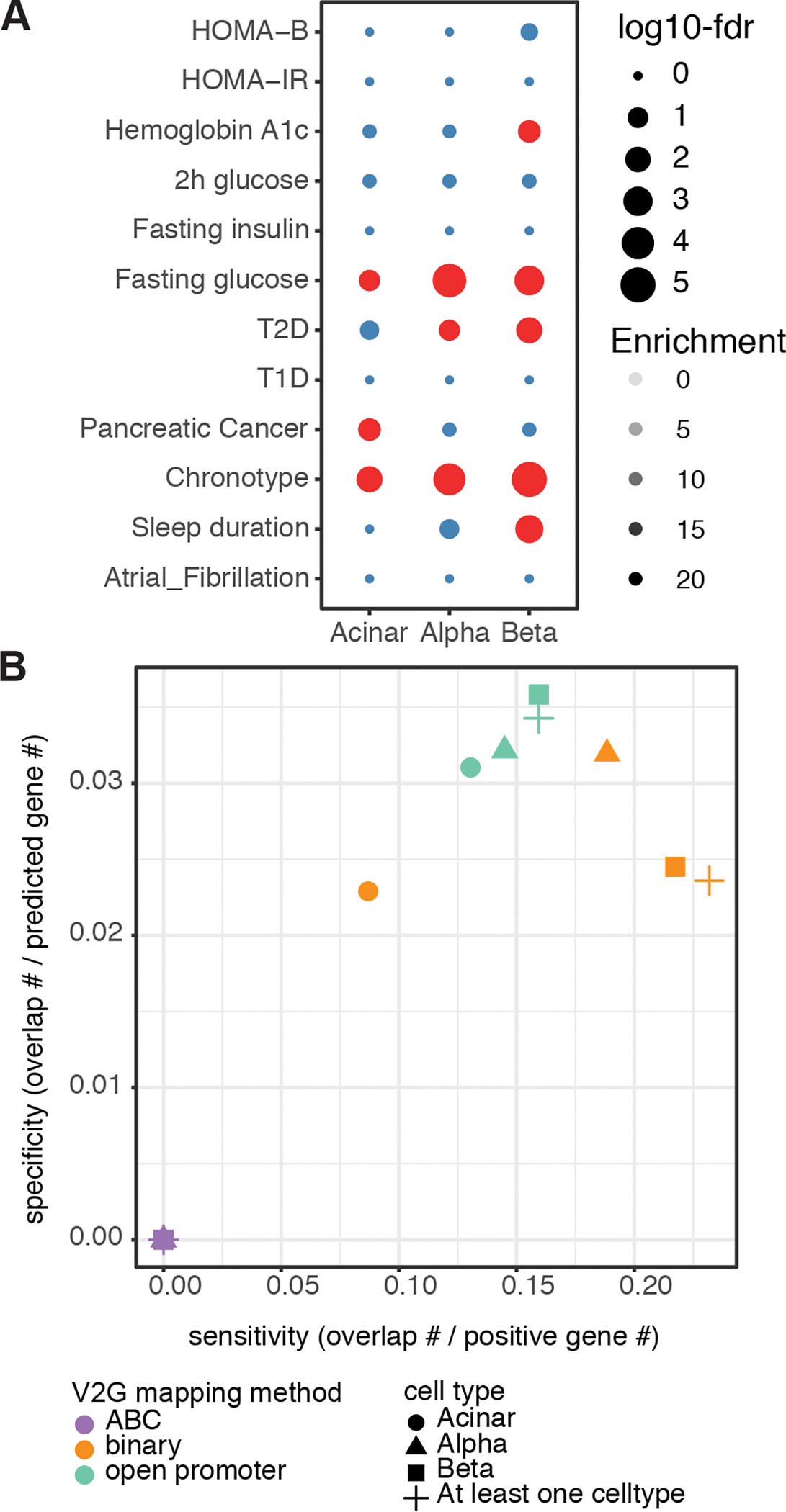
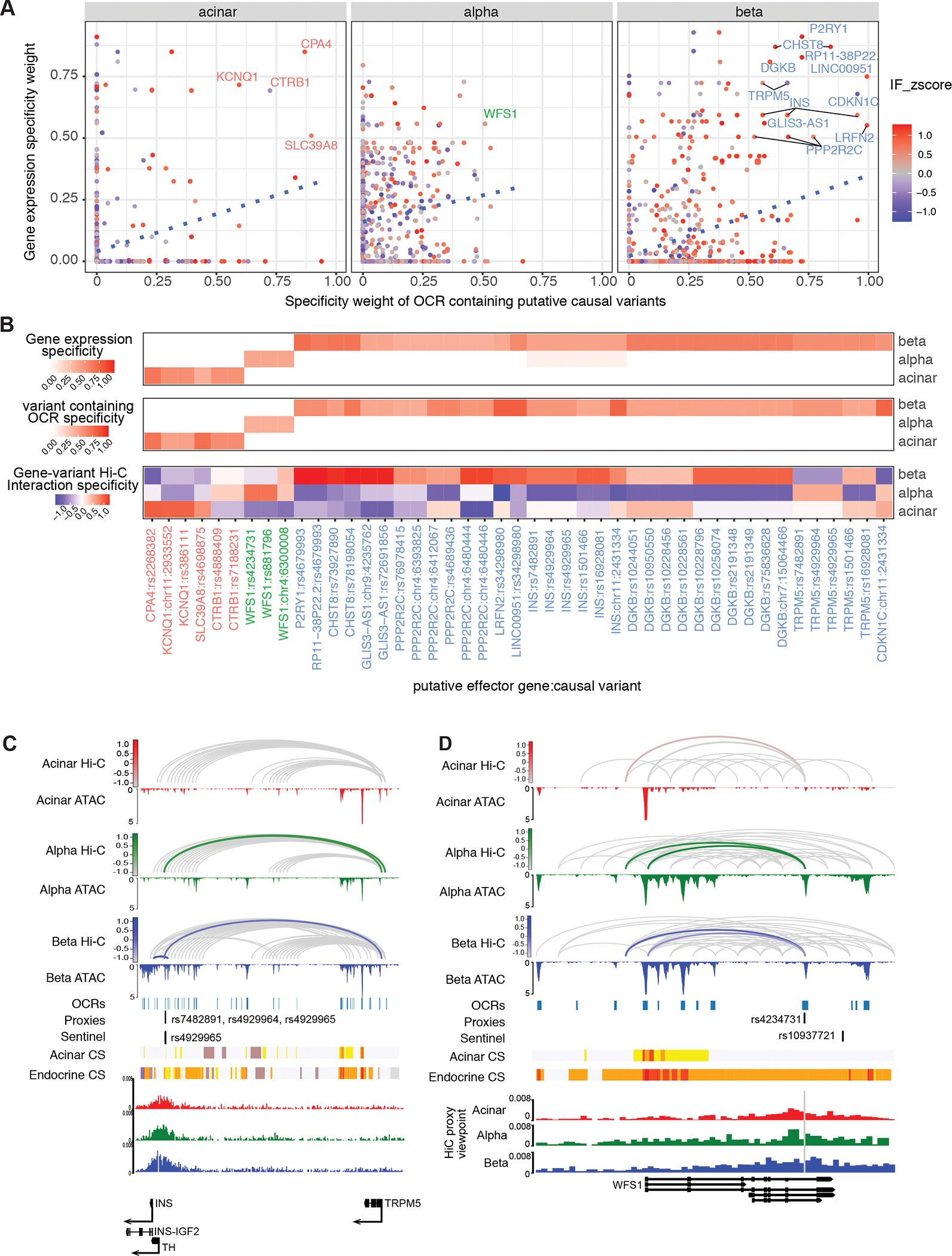
Three-dimensional (3D) chromatin organization maps help dissect cell-type-specific gene regulatory programs. Furthermore, 3D chromatin maps contribute to elucidating the pathogenesis of complex genetic diseases by connecting distal regulatory regions and genetic risk variants to their respective target genes. To understand the cell-type-specific regulatory architecture of diabetes risk, we generated transcriptomic and 3D epigenomic profiles of human pancreatic acinar, alpha, and beta cells using single-cell RNA-seq, single-cell ATAC-seq, and high-resolution Hi-C of sorted cells. Comparisons of these profiles revealed differential A/B (open/closed) chromatin compartmentalization, chromatin looping, and transcriptional factor-mediated control of cell-type-specific gene regulatory programs. We identified a total of 4,750 putative causal-variant-to-target-gene pairs at 194 type 2 diabetes GWAS signals using pancreatic 3D chromatin maps. We found that the connections between candidate causal variants and their putative target effector genes are cell-type stratified and emphasize previously underappreciated roles for alpha and acinar cells in diabetes pathogenesis.
Keywords: 3D chromatin maps; acinar cell; alpha cell; beta cell; islets of Langerhans; type 2 diabetes.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Alvarez-Dominguez JR, Donaghey J, Rasouli N, Kenty JHR, Helman A, Charlton J, Straubhaar JR, Meissner A, and Melton DA (2020). Circadian Entrainment Triggers Maturation of Human In Vitro Islets. Cell Stem Cell 26, 108–122 e110. - PubMed
-
- Beumer J, Puschhof J, Bauza-Martinez J, Martinez-Silgado A, Elmentaite R, James KR, Ross A, Hendriks D, Artegiani B, Busslinger GA, et al. (2020). High-Resolution mRNA and Secretome Atlas of Human Enteroendocrine Cells. Cell 182, 1062–1064. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
