

Integrated cohort of esophageal squamous cell cancer reveals genomic features underlying clinical characteristics
- PMID: 36071046
- PMCID: PMC9452532
- DOI: 10.1038/s41467-022-32962-1
Integrated cohort of esophageal squamous cell cancer reveals genomic features underlying clinical characteristics
Abstract
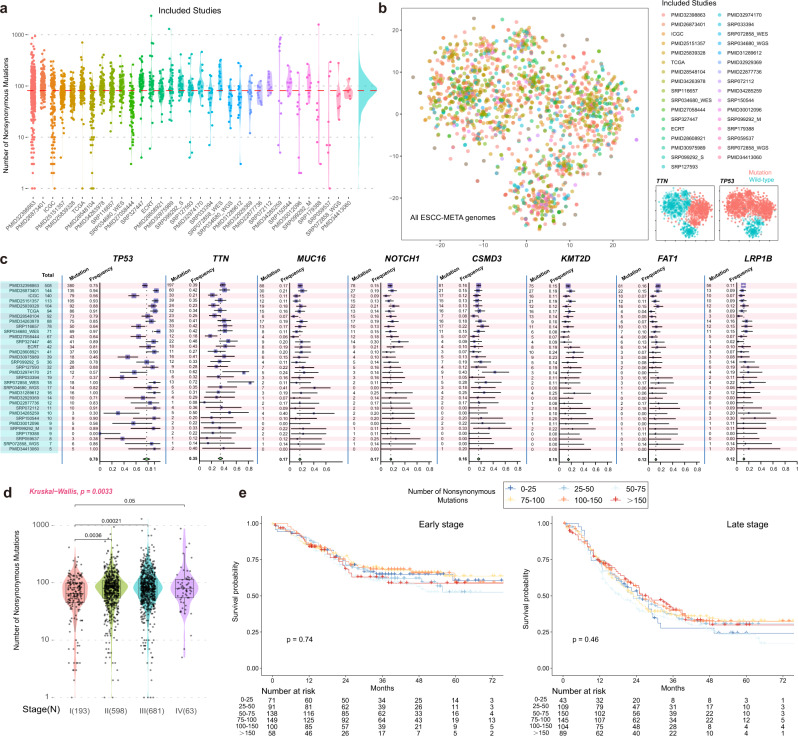
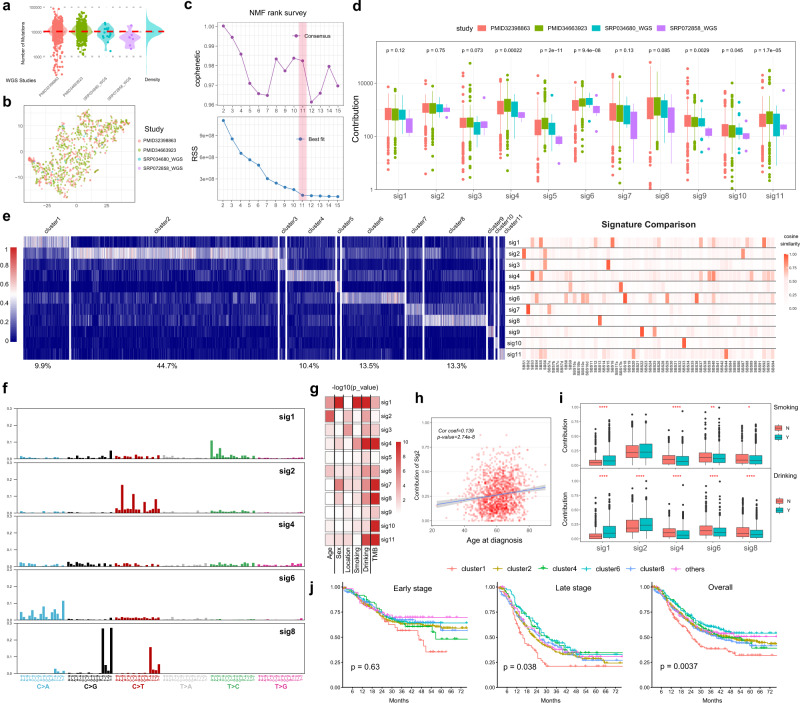
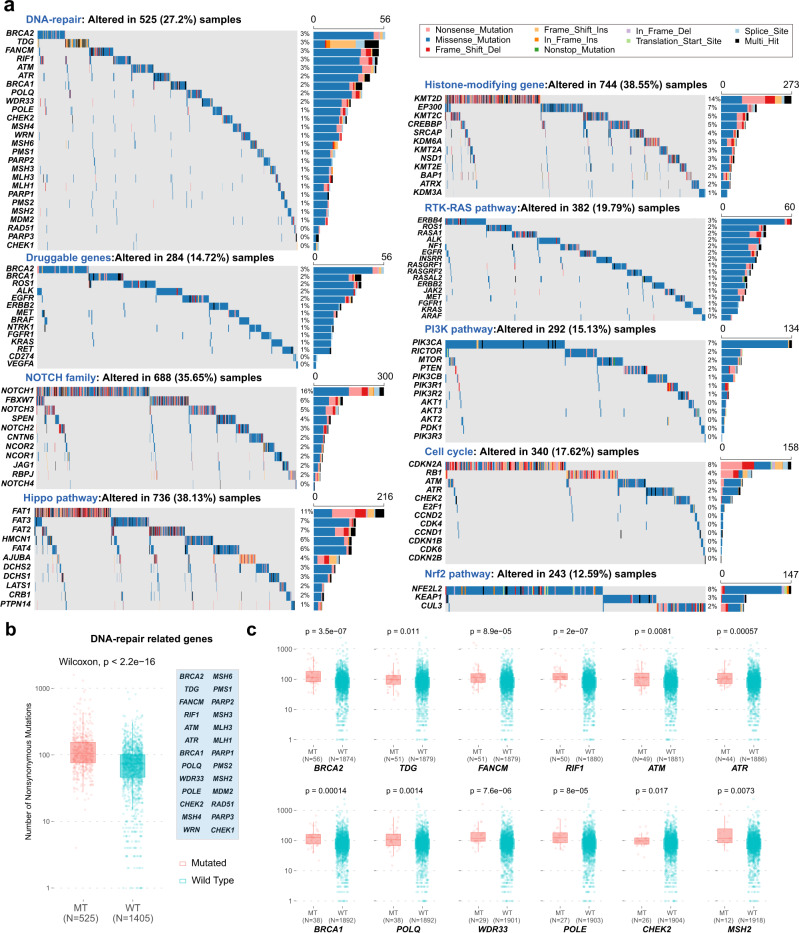
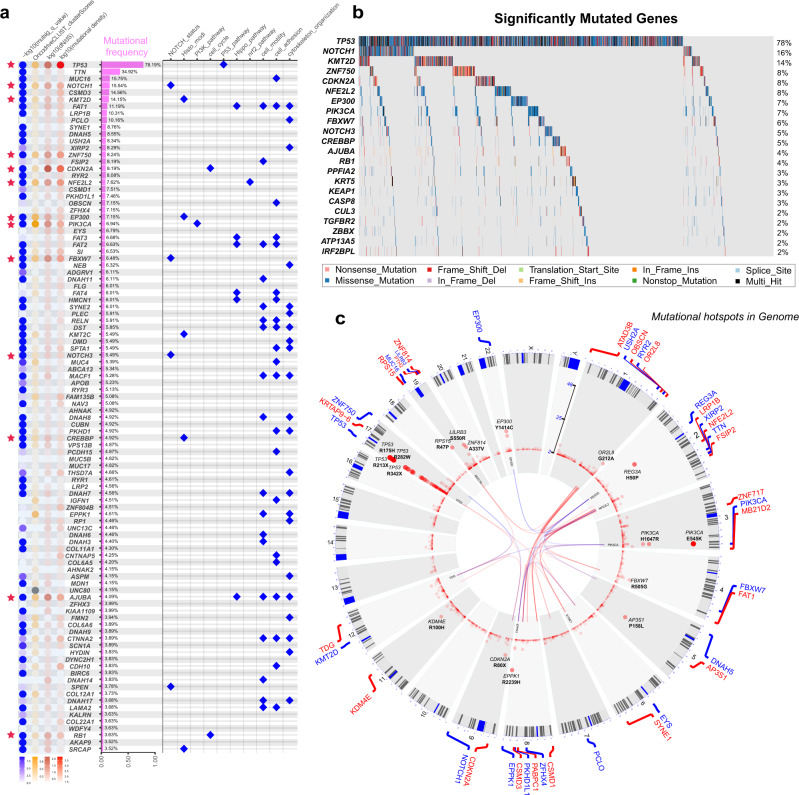
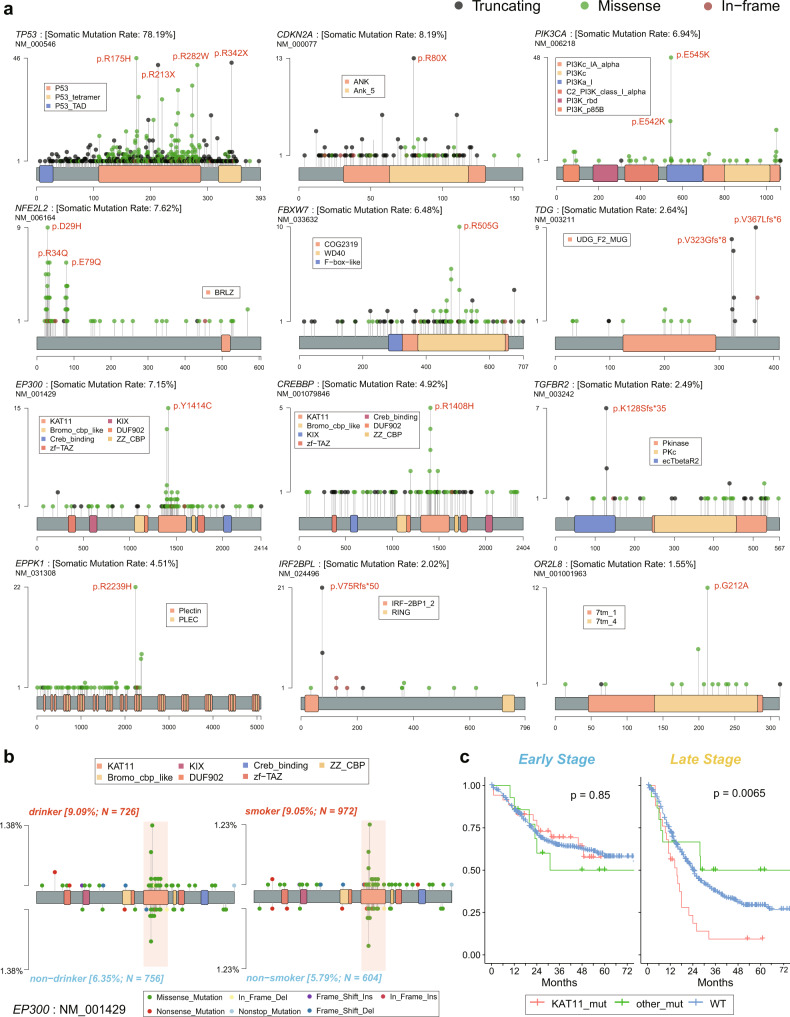
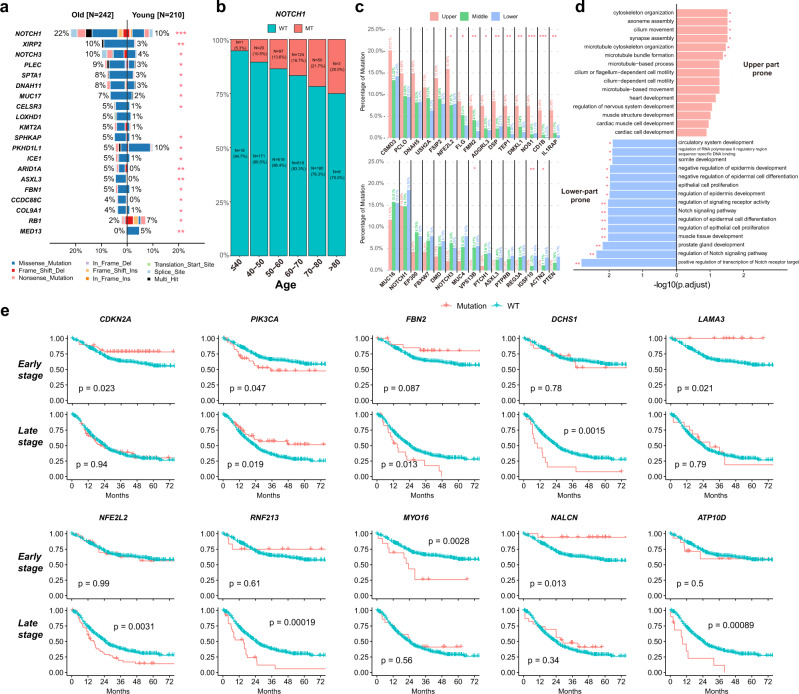
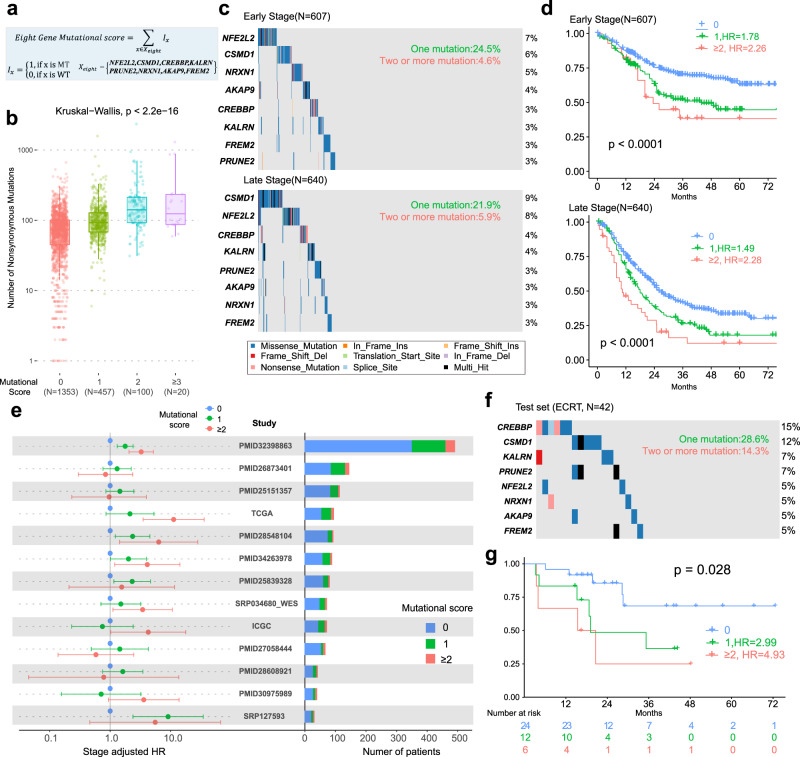
Esophageal squamous cell cancer (ESCC) is the major pathologic type of esophageal cancer in Asian population. To systematically evaluate the mutational features underlying clinical characteristics, we establish the integrated dataset of ESCC-META that consists of 1930 ESCC genomes from 33 datasets. The data process pipelines lead to well homogeneity of this integrated cohort for further analysis. We identified 11 mutational signatures in ESCC, some of which are related to clinical features, and firstly detect the significant mutated hotspots in TGFBR2 and IRF2BPL. We screen the survival related mutational features and found some genes had different prognostic impacts between early and late stage, such as PIK3CA and NFE2L2. Based on the results, an applicable approach of mutational score is proposed and validated to predict prognosis in ESCC. As an open-sourced, quality-controlled and updating mutational landscape, the ESCC-META dataset could facilitate further genomic and translational study in this field.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Agrawal N, et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012;2:899–905. doi: 10.1158/2159-8290.CD-12-0189. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
