

Targeting BCL-XL in fibrolamellar hepatocellular carcinoma
- PMID: 36073545
- PMCID: PMC9536265
- DOI: 10.1172/jci.insight.161820
Targeting BCL-XL in fibrolamellar hepatocellular carcinoma
Abstract
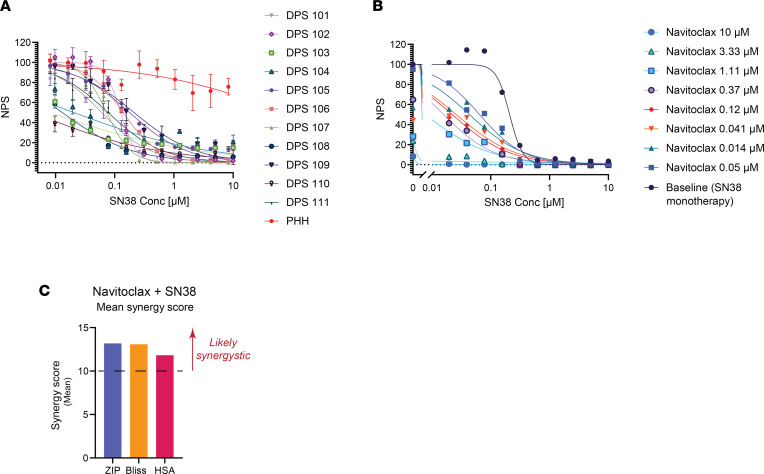
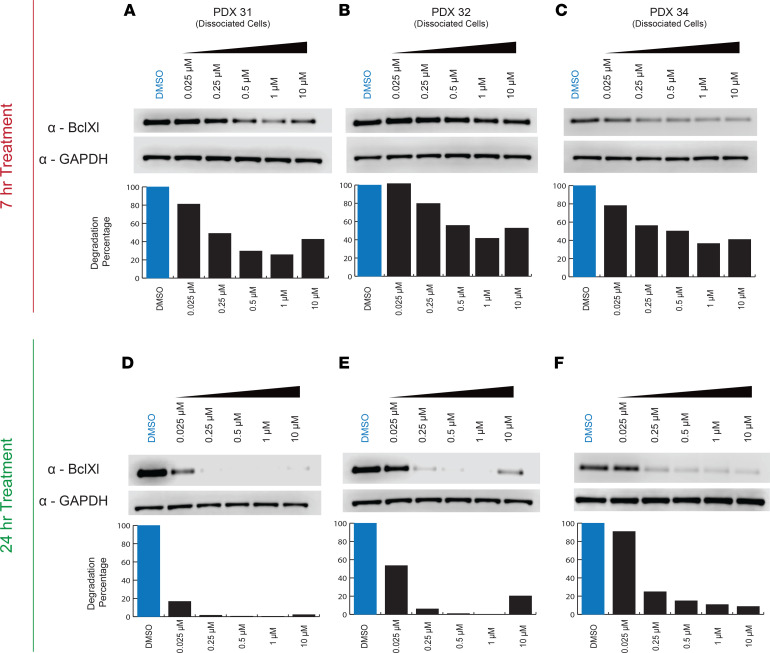
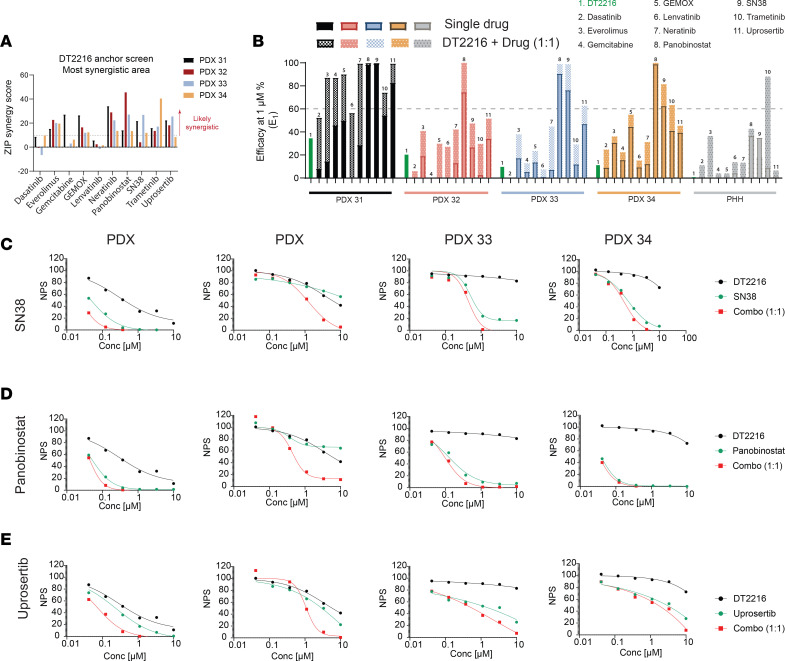
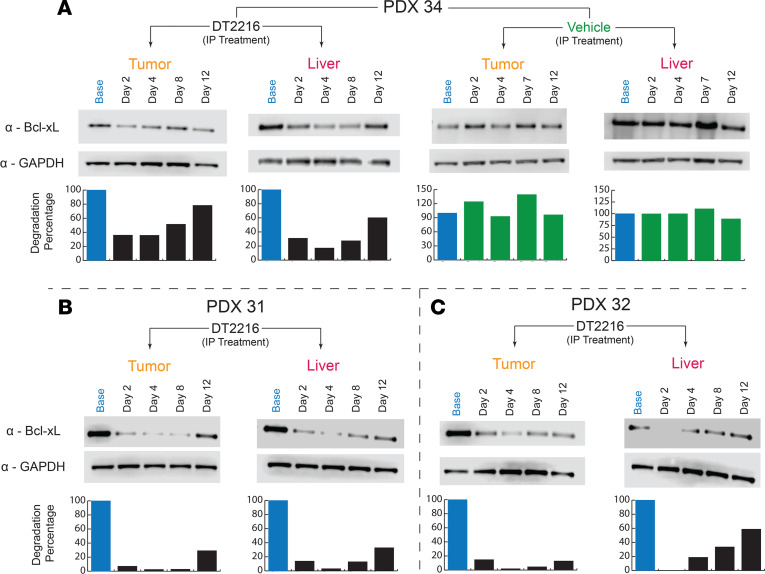
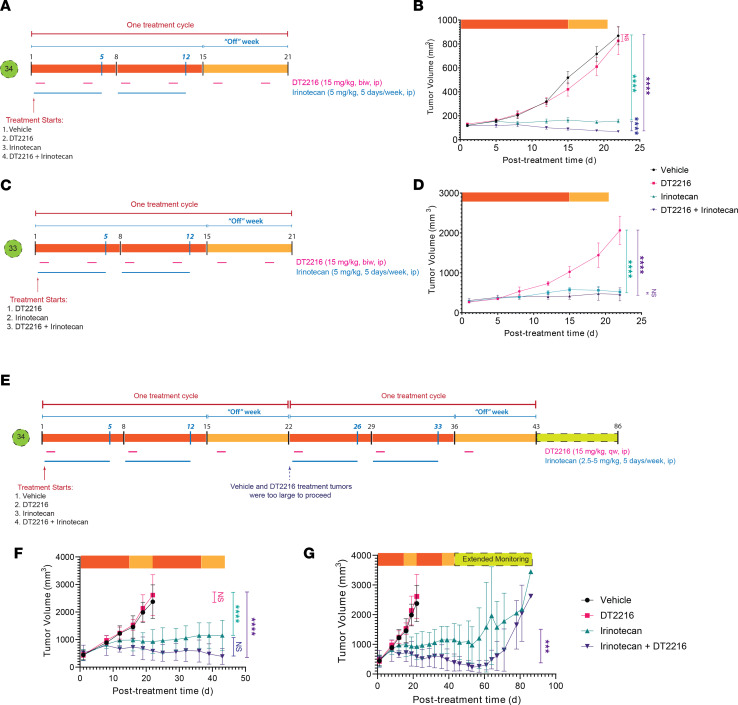
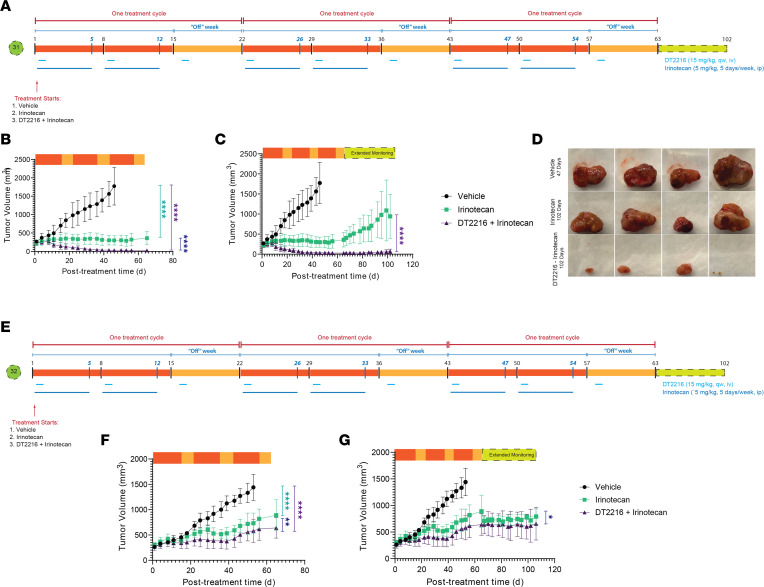
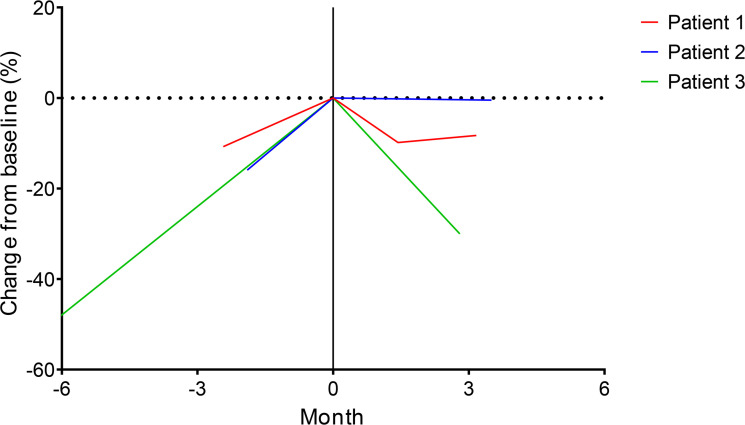
Fibrolamellar hepatocellular carcinoma (FLC) is a rare and often lethal liver cancer with no proven effective systemic therapy. Inhibition of the antiapoptotic protein BCL-XL was found to synergize with a variety of systemic therapies in vitro using cells dissociated from patient-derived xenografts (PDX) of FLC or cells dissociated directly from surgical patient resections. As BCL-XL is physiologically expressed in platelets, prior efforts to leverage this vulnerability in other cancers have been hampered by severe thrombocytopenia. To overcome this toxicity, we treated FLC models with DT2216, a proteolysis targeting chimera (PROTAC) that directs BCL-XL for degradation via the von Hippel-Lindau (VHL) E3 ligase, which is minimally expressed in platelets. The combination of irinotecan and DT2216 in vitro on cells directly acquired from patients or in vivo using several xenografts derived from patients with FLC demonstrated remarkable synergy and at clinically achievable doses not associated with significant thrombocytopenia.
Keywords: Apoptosis inhibitors; Drug therapy; Hepatology; Liver cancer; Oncology.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
