

Genomic Landscapes and Hallmarks of Mutant RAS in Human Cancers
- PMID: 36074020
- PMCID: PMC9627127
- DOI: 10.1158/0008-5472.CAN-22-1731
Genomic Landscapes and Hallmarks of Mutant RAS in Human Cancers
Abstract
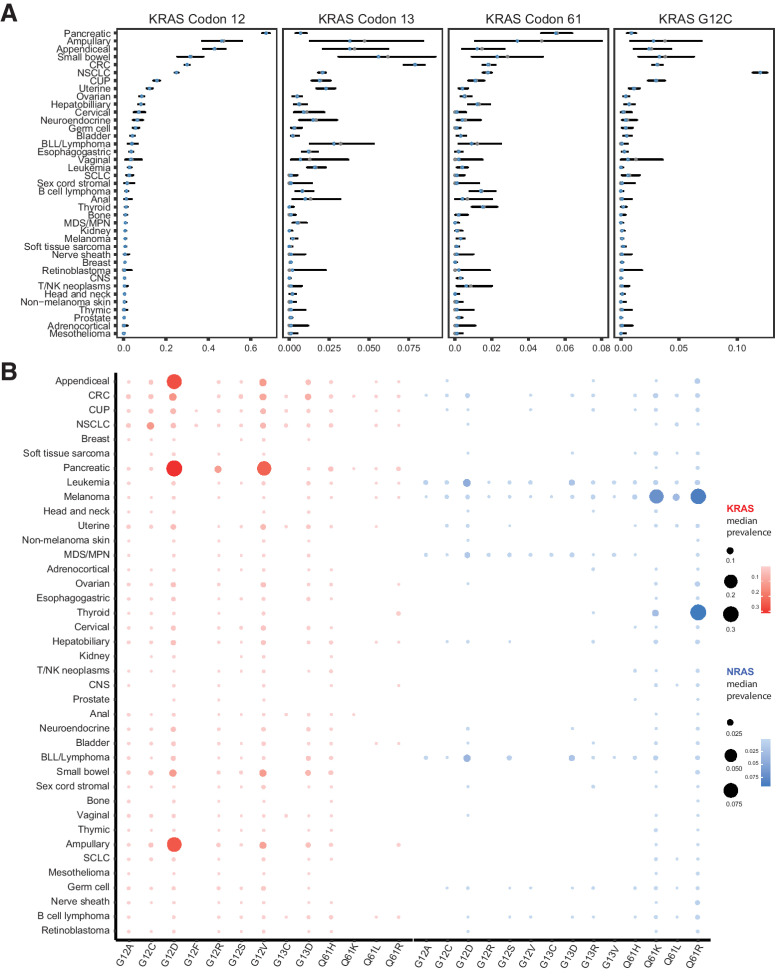
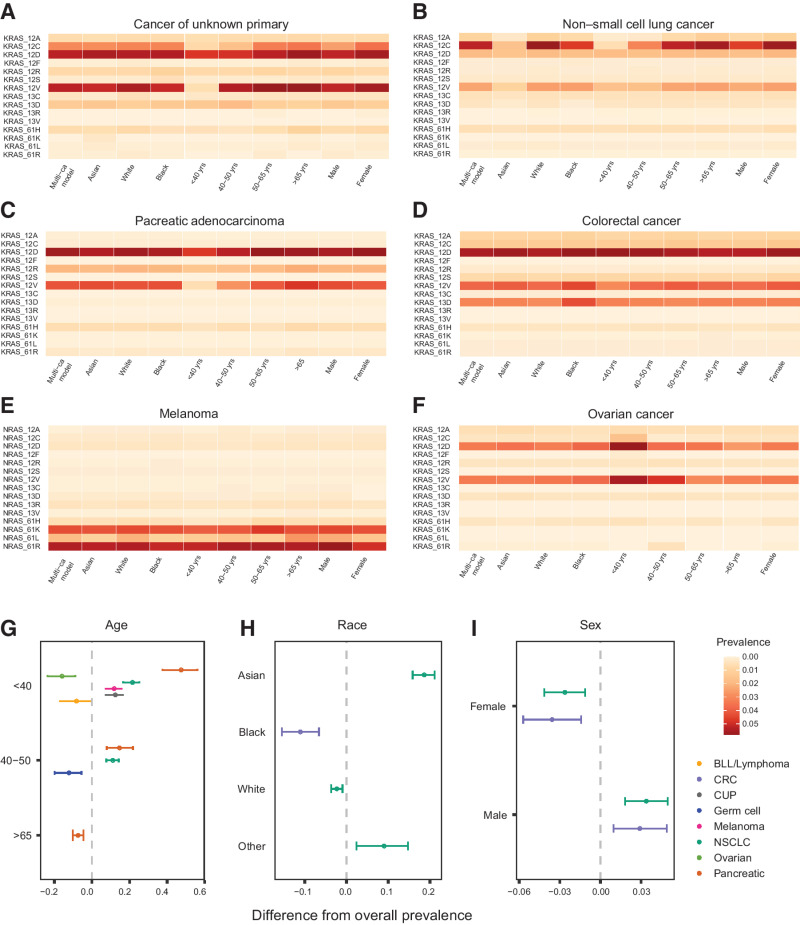
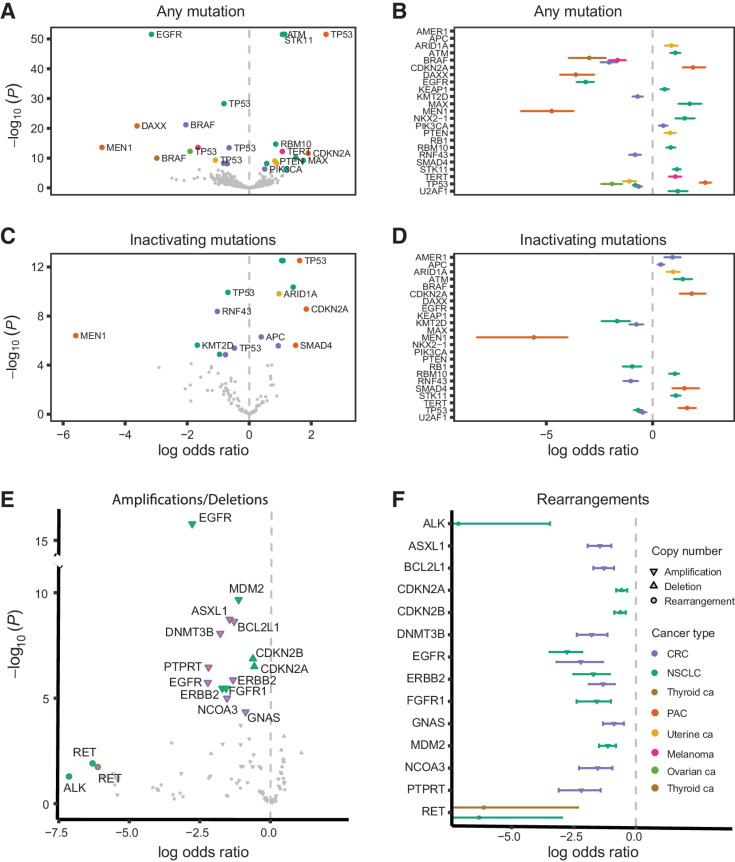
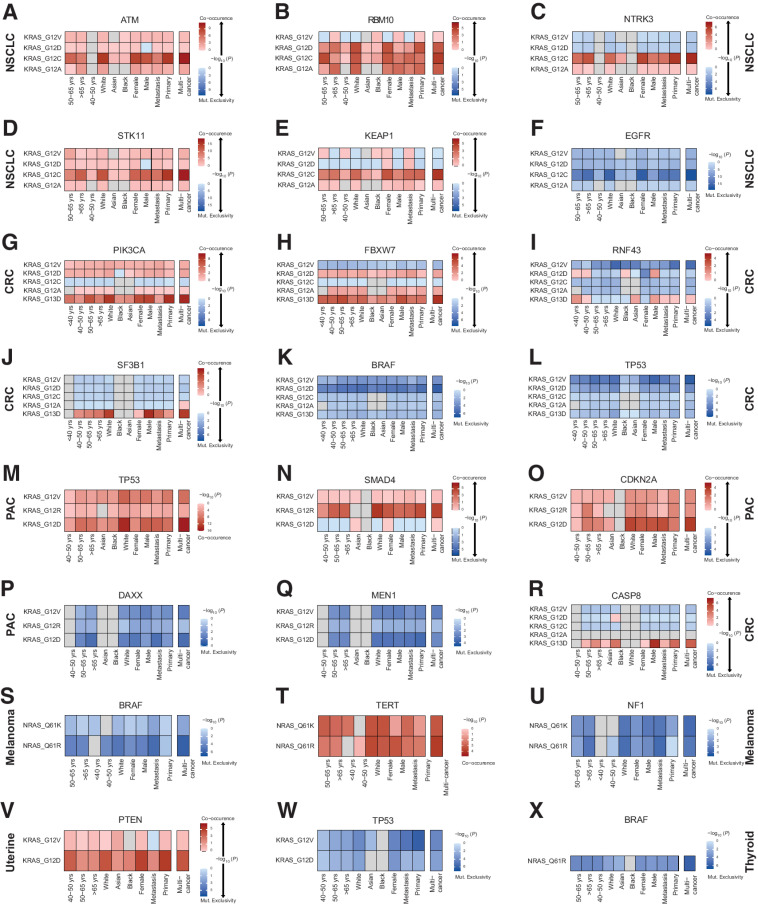
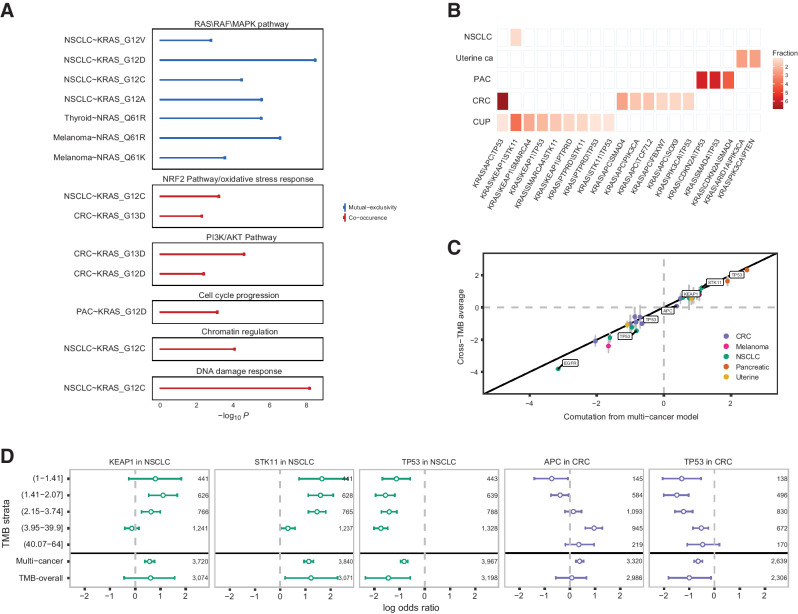
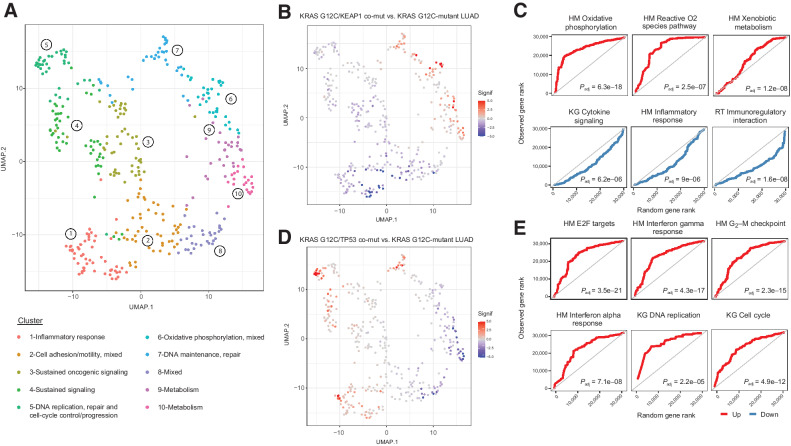
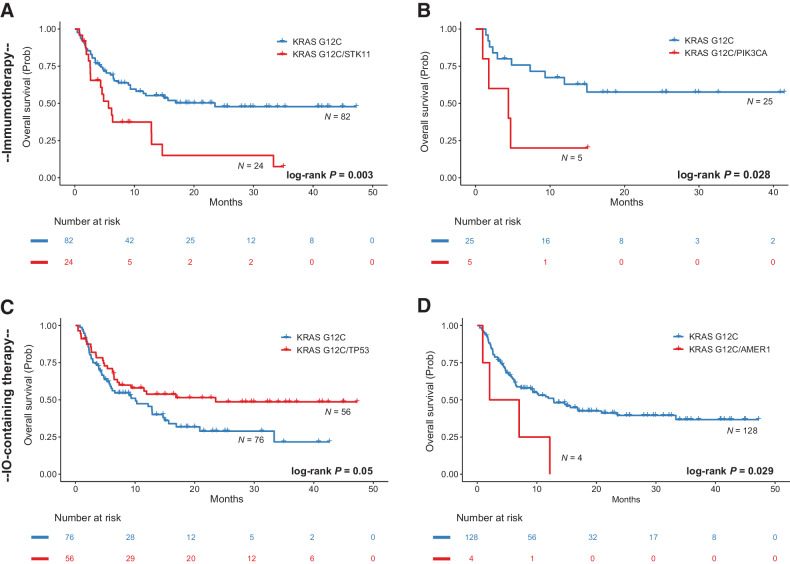
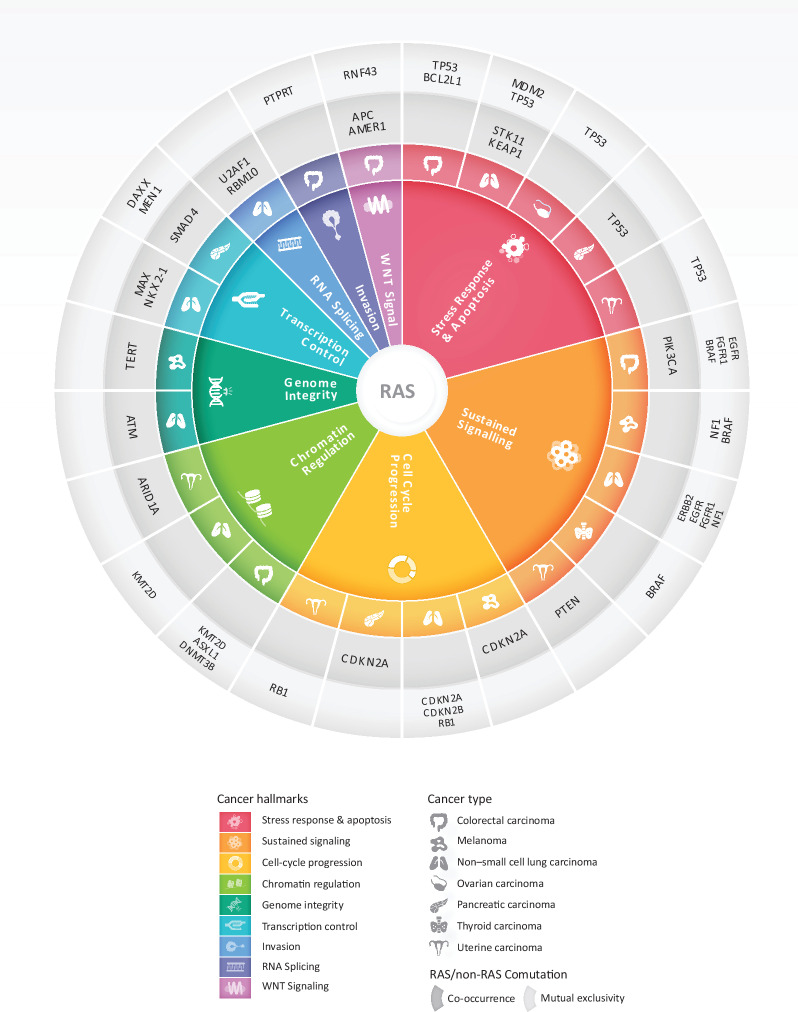
The RAS family of small GTPases represents the most commonly activated oncogenes in human cancers. To better understand the prevalence of somatic RAS mutations and the compendium of genes that are coaltered in RAS-mutant tumors, we analyzed targeted next-generation sequencing data of 607,863 mutations from 66,372 tumors in 51 cancer types in the AACR Project GENIE Registry. Bayesian hierarchical models were implemented to estimate the cancer-specific prevalence of RAS and non-RAS somatic mutations, to evaluate co-occurrence and mutual exclusivity, and to model the effects of tumor mutation burden and mutational signatures on comutation patterns. These analyses revealed differential RAS prevalence and comutations with non-RAS genes in a cancer lineage-dependent and context-dependent manner, with differences across age, sex, and ethnic groups. Allele-specific RAS co-mutational patterns included an enrichment in NTRK3 and chromatin-regulating gene mutations in KRAS G12C-mutant non-small cell lung cancer. Integrated multiomic analyses of 10,217 tumors from The Cancer Genome Atlas (TCGA) revealed distinct genotype-driven gene expression programs pointing to differential recruitment of cancer hallmarks as well as phenotypic differences and immune surveillance states in the tumor microenvironment of RAS-mutant tumors. The distinct genomic tracks discovered in RAS-mutant tumors reflected differential clinical outcomes in TCGA cohort and in an independent cohort of patients with KRAS G12C-mutant non-small cell lung cancer that received immunotherapy-containing regimens. The RAS genetic architecture points to cancer lineage-specific therapeutic vulnerabilities that can be leveraged for rationally combining RAS-mutant allele-directed therapies with targeted therapies and immunotherapy.
Significance: The complex genomic landscape of RAS-mutant tumors is reflective of selection processes in a cancer lineage-specific and context-dependent manner, highlighting differential therapeutic vulnerabilities that can be clinically translated.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res 2015;13:1325–35. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
