

Cancer genome and tumor microenvironment: Reciprocal crosstalk shapes lung cancer plasticity
- PMID: 36074553
- PMCID: PMC9457687
- DOI: 10.7554/eLife.79895
Cancer genome and tumor microenvironment: Reciprocal crosstalk shapes lung cancer plasticity
Abstract
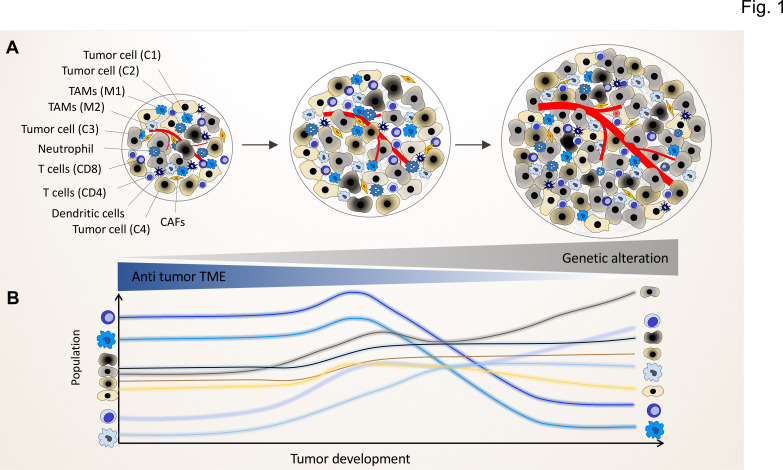
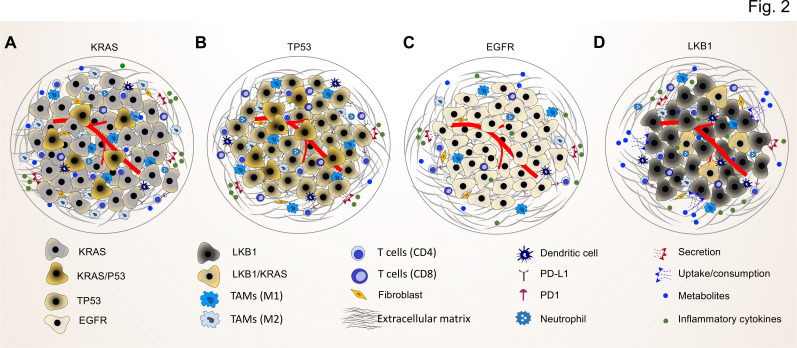
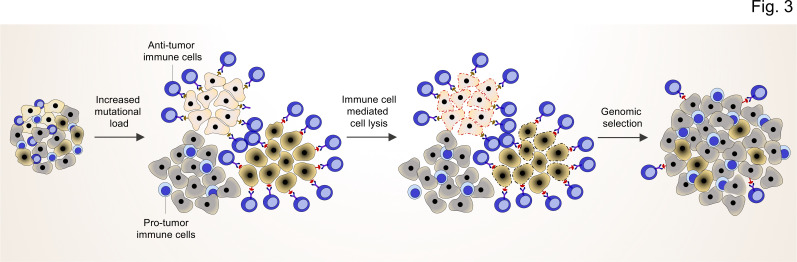
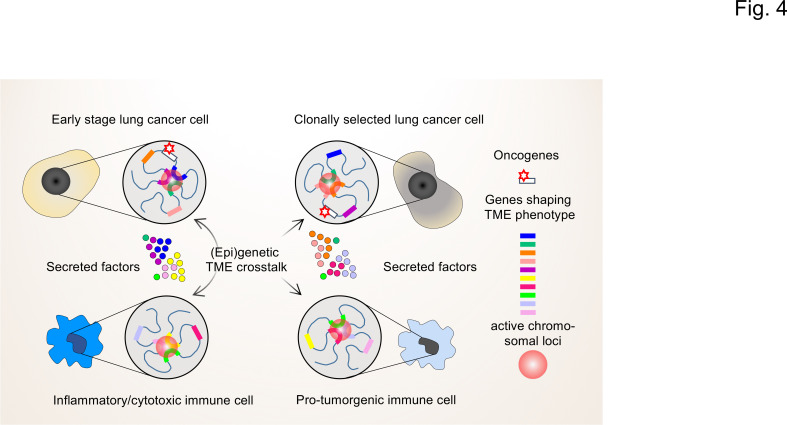
Lung cancer classification and treatment has been revolutionized by improving our understanding of driver mutations and the introduction of tumor microenvironment (TME)-associated immune checkpoint inhibitors. Despite the significant improvement of lung cancer patient survival in response to either oncogene-targeted therapy or anticancer immunotherapy, many patients show initial or acquired resistance to these new therapies. Recent advances in genome sequencing reveal that specific driver mutations favor the development of an immunosuppressive TME phenotype, which may result in unfavorable outcomes in lung cancer patients receiving immunotherapies. Clinical studies with follow-up after immunotherapy, assessing oncogenic driver mutations and the TME immune profile, not only reveal the underlying potential molecular mechanisms in the resistant lung cancer patients but also hold the key to better treatment choices and the future of personalized medicine. In this review, we discuss the crosstalk between cancer cell genomic features and the TME to reveal the impact of genetic alterations on the TME phenotype. We also provide insights into the regulatory role of cellular TME components in defining the genetic landscape of cancer cells during tumor development.
Keywords: cancer biology; cancer genome; immunology; inflammation; lung cancer; tumor microenvironment.
© 2022, Mansouri et al.
Conflict of interest statement
SM, DH, TS, MK, RS No competing interests declared
Figures
References
-
- Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, Wilkerson MD, Fecci PE, Butaney M, Reibel JB, Soucheray M, Cohoon TJ, Janne PA, Meyerson M, Hayes DN, Shapiro GI, Shimamura T, Sholl LM, Rodig SJ, Freeman GJ, Hammerman PS, Dranoff G, Wong K-K. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discovery. 2013;3:1355–1363. doi: 10.1158/2159-8290.CD-13-0310. - DOI - PMC - PubMed
-
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Imielinsk M, Jäger N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van ’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain. Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
