

Diagnosis and treatment of thrombotic microangiopathy
- PMID: 36074708
- PMCID: PMC9544907
- DOI: 10.1111/ijlh.13954
Diagnosis and treatment of thrombotic microangiopathy
Abstract
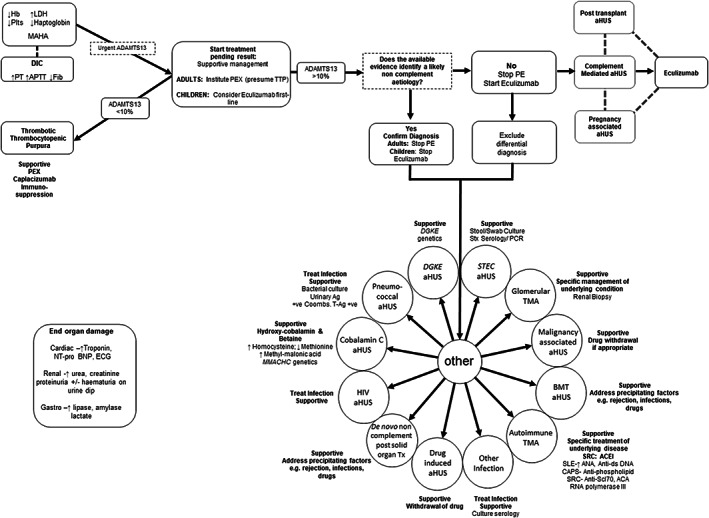
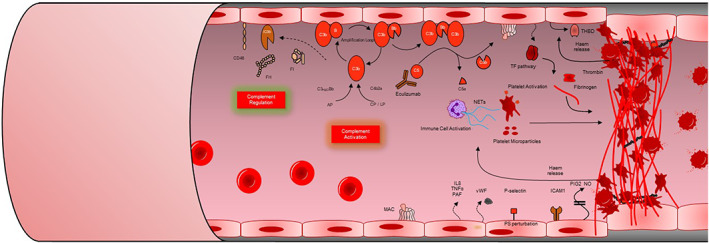
Thrombotic microangiopathy (TMA) is characterized by thrombocytopenia, microangiopathic haemolytic anaemia and end organ damage. TMAs have varying underlying pathophysiology and can therefore present with an array of clinical presentations. Renal involvement is common as the kidney is particularly susceptible to the endothelial damage and microvascular occlusion. TMAs require rapid assessment, diagnosis, and commencement of appropriate treatment due to the high morbidity and mortality associated with them. Ground-breaking research into the pathogenesis of TMAs over the past 20 years has driven the successful development of targeted therapeutics revolutionizing patient outcomes. This review outlines the clinical presentations, pathogenesis, diagnostic tests and treatments for TMAs.
Keywords: STEC-HUS; haemolytic uraemic syndrome; thrombotic microangiopathy; thrombotic thrombocytopenic purpura.
© 2022 The Authors. International Journal of Laboratory Hematology published by John Wiley & Sons Ltd.
Conflict of interest statement
David Kavanagh has received consultancy income from Gyroscope Therapeutics, Silence Therapeutics, Alexion Pharmaceuticals, Novartis, Apellis, and Sarepta.
Figures
References
-
- Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347(8):589‐600. - PubMed
-
- Scully M, Yarranton H, Liesner R, et al. Regional UK TTP registry: correlation with laboratory ADAMTS 13 analysis and clinical features. Br J Haematol. 2008;142(5):819‐826. - PubMed
-
- Alwan F, Vendramin C, Vanhoorelbeke K, et al. Presenting ADAMTS13 antibody and antigen levels predict prognosis in immune‐mediated thrombotic thrombocytopenic purpura. Blood. 2017;130(4):466‐471. - PubMed
-
- Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017;129(21):2836‐2846. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
