

The hemoglobinopathies, molecular disease mechanisms and diagnostics
- PMID: 36074711
- PMCID: PMC9542123
- DOI: 10.1111/ijlh.13885
The hemoglobinopathies, molecular disease mechanisms and diagnostics
Abstract
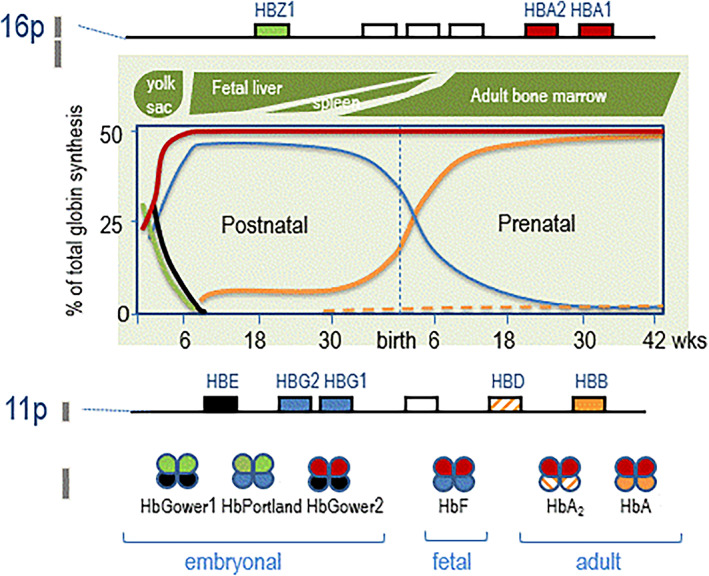
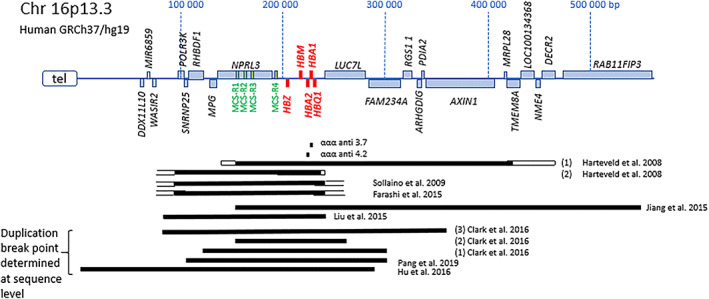
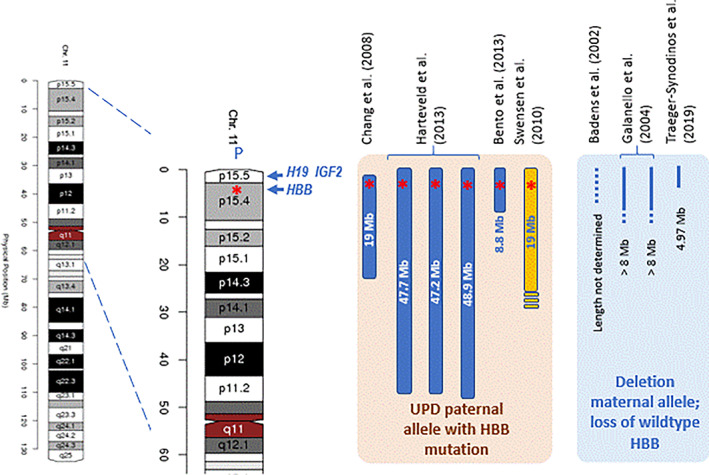
Hemoglobinopathies are the most common monogenic disorders in the world with an ever increasing global disease burden each year. As most hemoglobinopathies show recessive inheritance carriers are usually clinically silent. Programmes for preconception and antenatal carrier screening, with the option of prenatal diagnosis are considered beneficial in many endemic countries. With the development of genetic tools such as Array analysis and Next Generation Sequencing in addition to state of the art screening at the hematologic, biochemic and genetic level, have contributed to the discovery of an increasing number of rare rearrangements and novel factors influencing the disease severity over the recent years. This review summarizes the basic requirements for adequate carrier screening analysis, the importance of genotype-phenotype correlation and how this may lead to the unrevealing exceptional interactions causing a clinically more severe phenotype in otherwise asymptomatic carriers. A special group of patients are β-thalassemia carriers presenting with features of β-thalassemia intermedia of various clinical severity. The disease mechanisms may involve duplicated α-globin genes, mosaic partial Uniparental Isodisomy of chromosome 11p15.4 where the HBB gene is located or haplo-insufficiency of a non-linked gene SUPT5H on chromosome 19q, first described in two Dutch families with β-thalassemia trait without variants in the HBB gene.
Keywords: haematology; haemoglobin; molecular diagnosis; sickle cell disease; thalassemia; β-thalassemia intermedia.
© 2022 The Authors. International Journal of Laboratory Hematology published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Steinberg MH, Forget BG, Higgs DR, Nagel RL. Disorders of Hemoglobin. 2nd ed. Genetics, Pathophysiology and Clinical Management; 2009.
-
- Haldane JB. The rate of mutation of human genes. Hereditas. 1949;35:267‐273.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
