

Neuroinflammation represents a common theme amongst genetic and environmental risk factors for Alzheimer and Parkinson diseases
- PMID: 36076238
- PMCID: PMC9452283
- DOI: 10.1186/s12974-022-02584-x
Neuroinflammation represents a common theme amongst genetic and environmental risk factors for Alzheimer and Parkinson diseases
Abstract
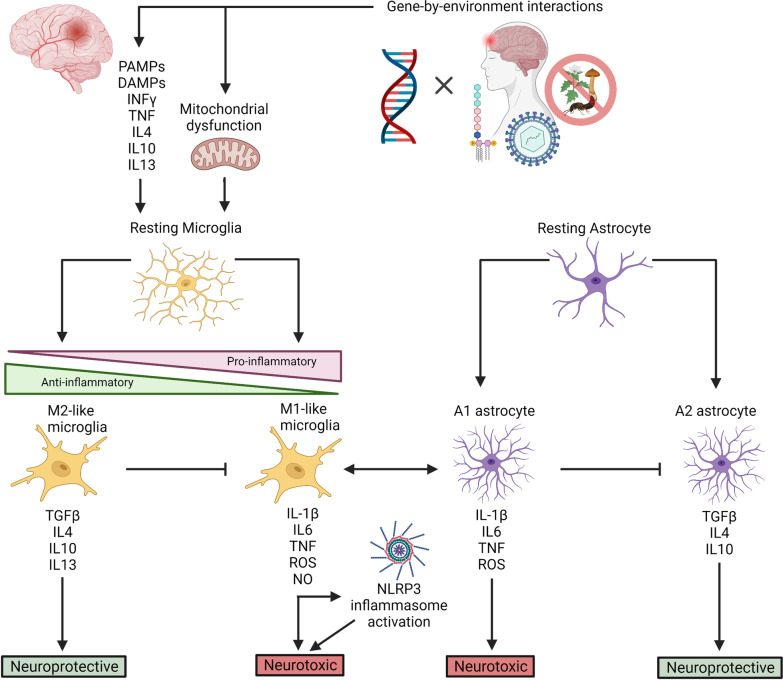
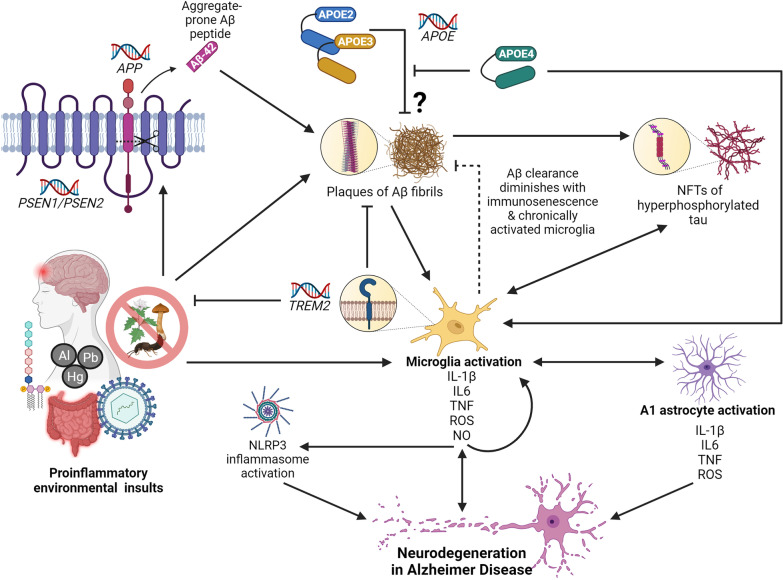
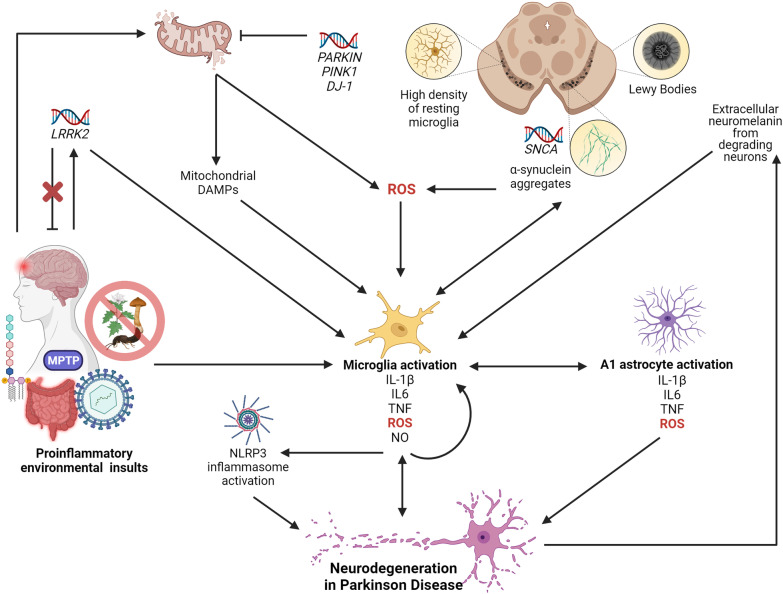
Multifactorial diseases are characterized by inter-individual variation in etiology, age of onset, and penetrance. These diseases tend to be relatively common and arise from the combined action of genetic and environmental factors; however, parsing the convoluted mechanisms underlying these gene-by-environment interactions presents a significant challenge to their study and management. For neurodegenerative disorders, resolving this challenge is imperative, given the enormous health and societal burdens they impose. The mechanisms by which genetic and environmental effects may act in concert to destabilize homeostasis and elevate risk has become a major research focus in the study of common disease. Emphasis is further being placed on determining the extent to which a unifying biological principle may account for the progressively diminishing capacity of a system to buffer disease phenotypes, as risk for disease increases. Data emerging from studies of common, neurodegenerative diseases are providing insights to pragmatically connect mechanisms of genetic and environmental risk that previously seemed disparate. In this review, we discuss evidence positing inflammation as a unifying biological principle of homeostatic destabilization affecting the risk, onset, and progression of neurodegenerative diseases. Specifically, we discuss how genetic variation associated with Alzheimer disease and Parkinson disease may contribute to pro-inflammatory responses, how such underlying predisposition may be exacerbated by environmental insults, and how this common theme is being leveraged in the ongoing search for effective therapeutic interventions.
Keywords: Alzheimer disease; Chronic inflammation; Common genetic disease; Gene-by-environment interactions; Inflammatory response; Multifactorial phenotypes; Neurodegeneration; Neuroinflammation; Parkinson disease.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–1102. doi: 10.1016/S1474-4422(19)30320-5. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
