

Diverse Effects of Cilostazol on Proprotein Convertase Subtilisin/Kexin Type 9 between Obesity and Non-Obesity
- PMID: 36077166
- PMCID: PMC9456424
- DOI: 10.3390/ijms23179768
Diverse Effects of Cilostazol on Proprotein Convertase Subtilisin/Kexin Type 9 between Obesity and Non-Obesity
Abstract
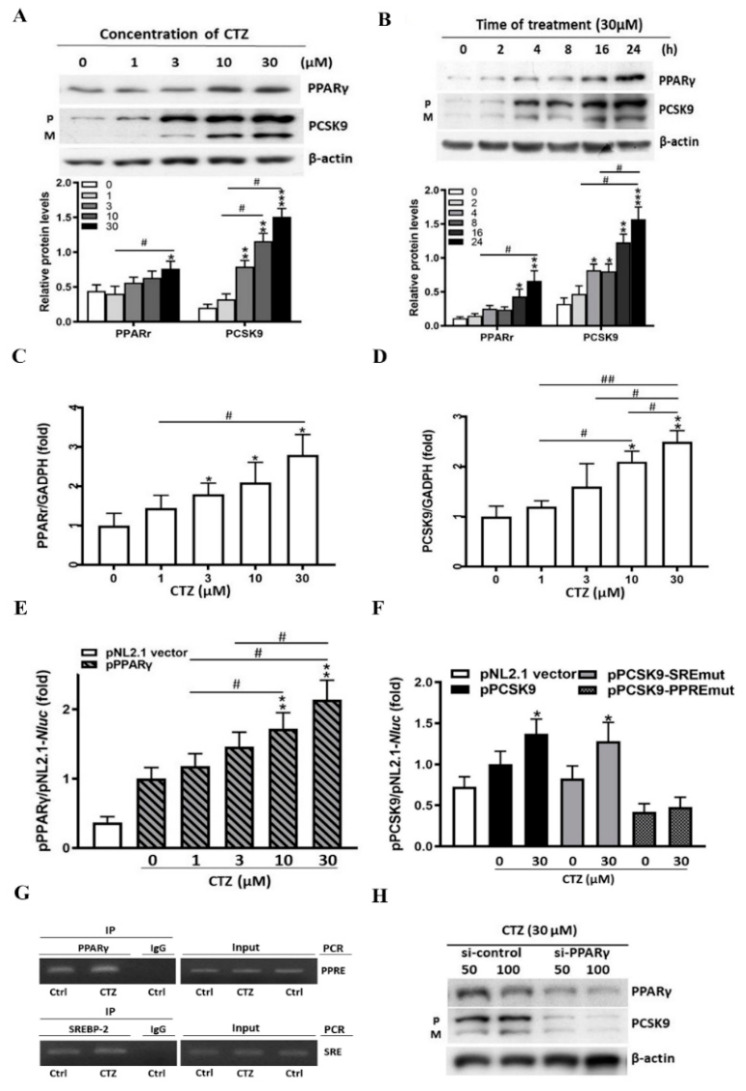
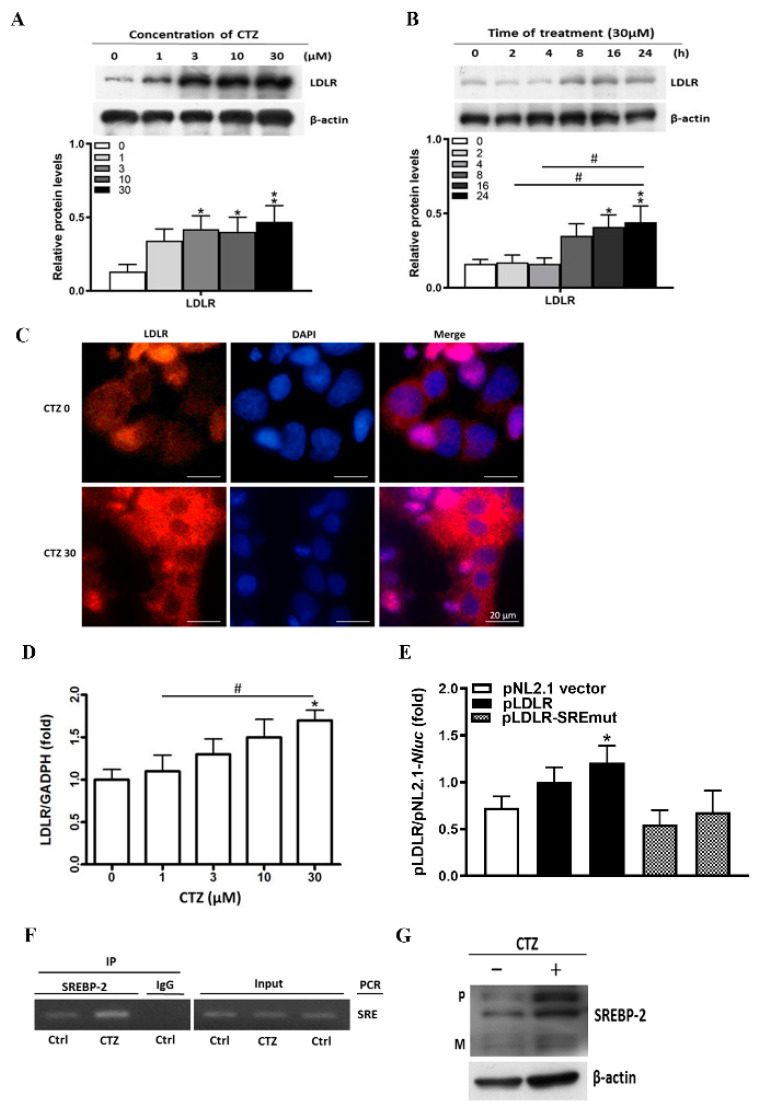
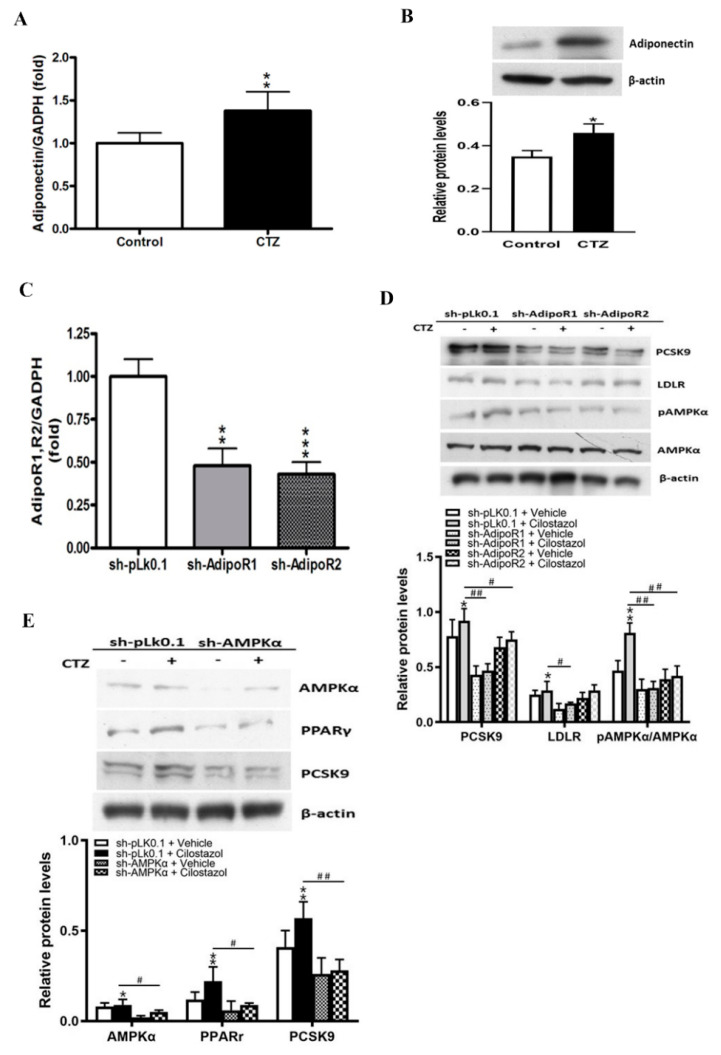
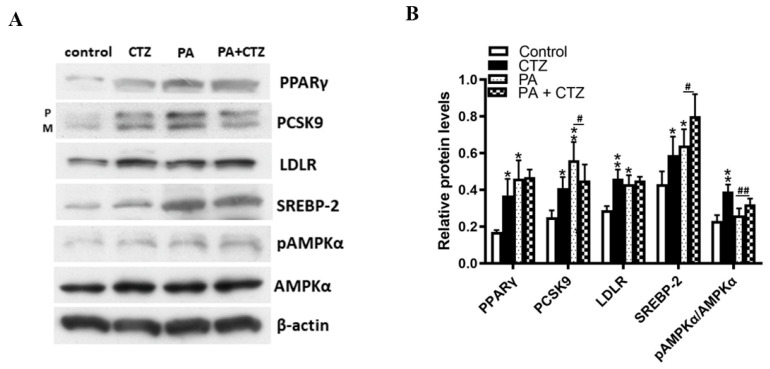
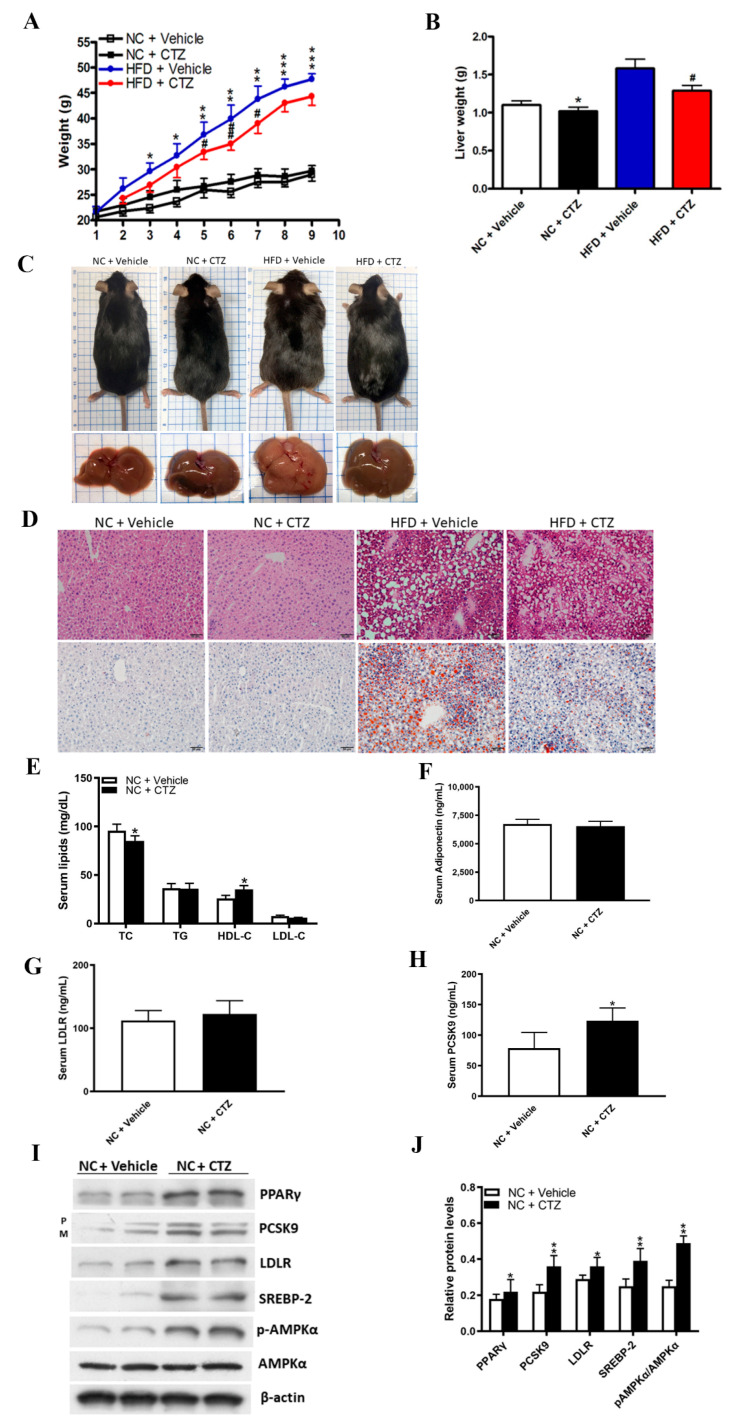
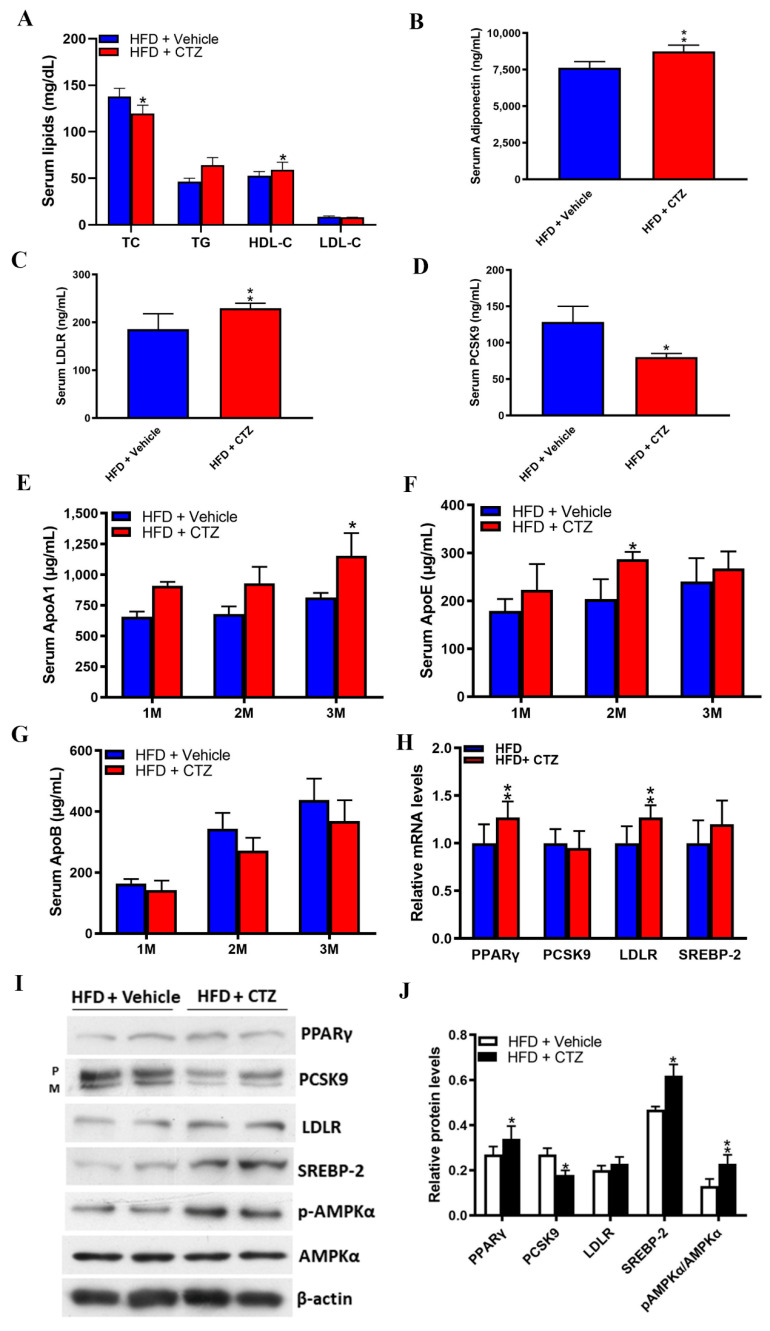
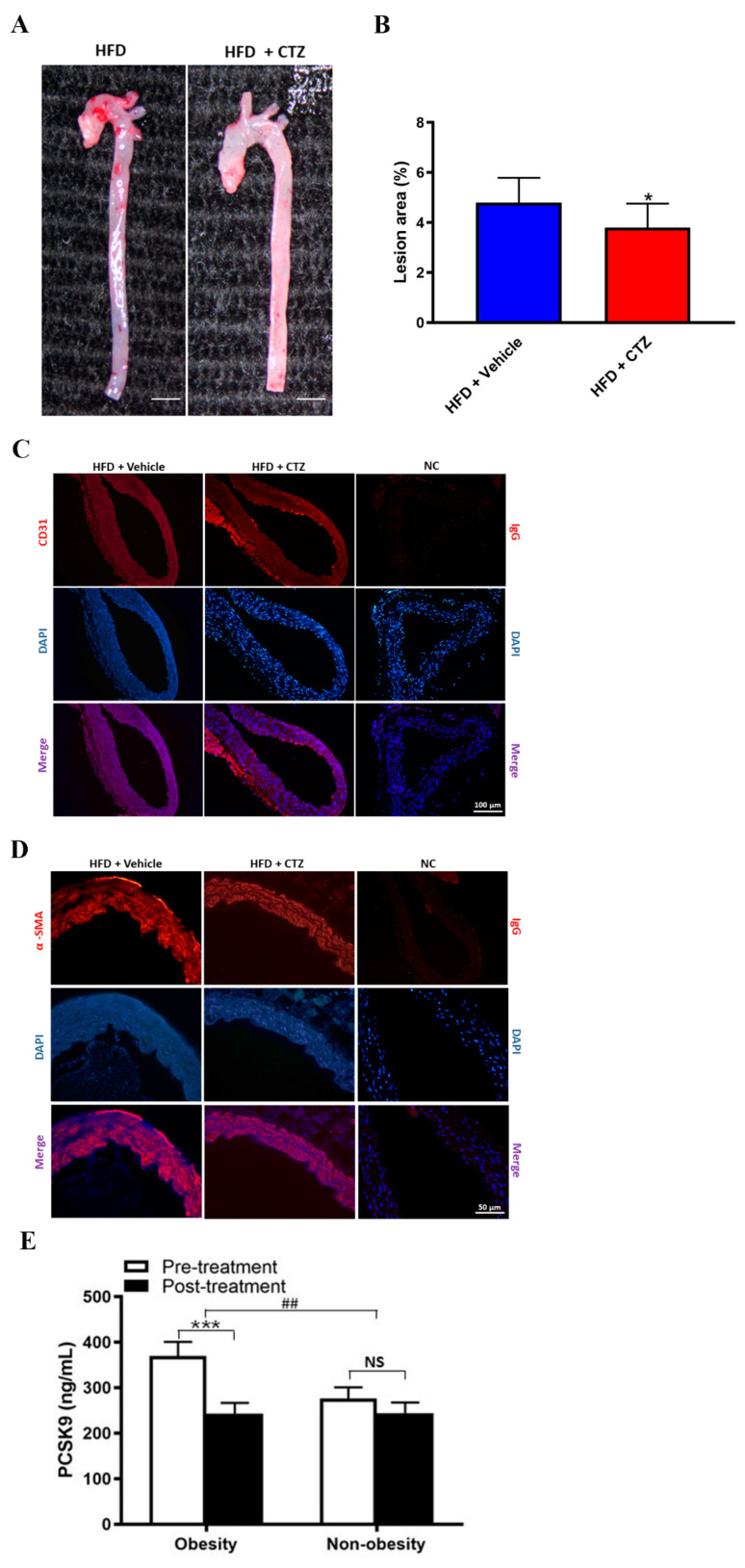
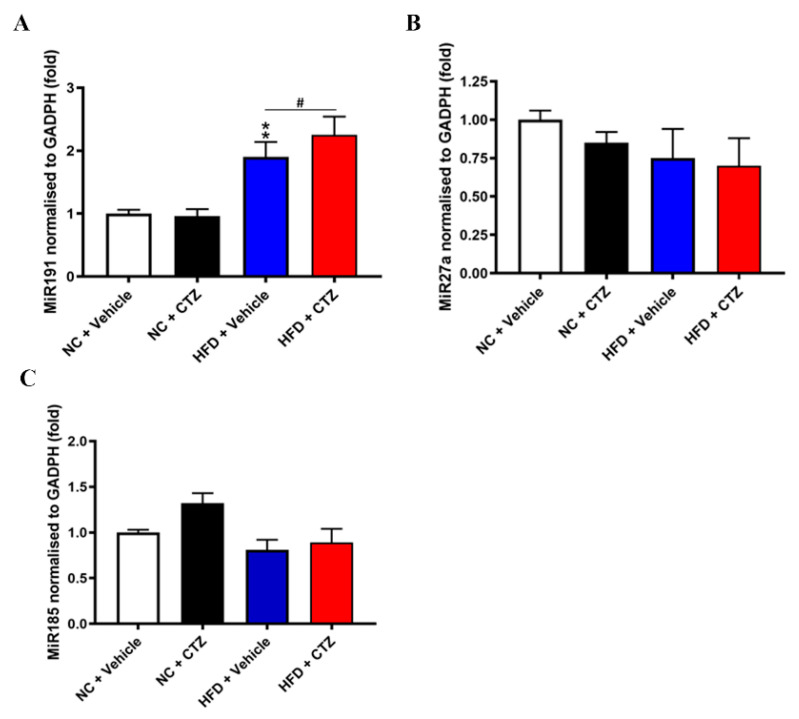
Proprotein convertase subtilisin/kexin type 9 (PCSK9) plays a key role in cholesterol homeostasis. Cilostazol exerts favorable cellular and metabolic effects; however, the effect of cilostazol on the expression of PCSK9 has not been previously reported. Our study aimed to investigate the potential mechanisms of action of cilostazol on the expression of PCSK9 and lipid homeostasis. We evaluated the effects of cilostazol on the expression of PCSK9 in HepG2 cells and evaluated potential molecular mechanisms by measuring signaling molecules in the liver and serum lipid profiles in high-fat diet-induced obese mice and normal chow-fed mice. Cilostazol treatment significantly induced the messenger RNA and protein expression of PCSK9 in HepG2 cells and enhanced PCSK9 promoter activity. Chromatin immunoprecipitation assays confirmed that cilostazol treatment enhanced PCSK9 transcription by binding to peroxisome proliferator-activated receptor-γ (PPARγ) via the PPARγ DNA response element. PPARγ knockdown attenuated the stimulatory effect of cilostazol on PCSK9. In vitro, cilostazol treatment increased PCSK9 expression in vehicle-treated HepG2 cells but decreased PCSK9 expression in palmitic acid-treated HepG2 cells. In vivo, cilostazol treatment increased the serum levels of PCSK9 in normal mice but significantly reduced PCSK9 levels in obese mice. The expressions of PCSK9-relevant microRNAs also showed similar results. Clinical data showed that cilostazol treatment significantly reduced serum PCSK9 levels in patients with obesity. The obesity-dependent effects of cilostazol on PCSK9 expression observed from bench to bedside demonstrates the therapeutic potential of cilostazol in clinical settings.
Keywords: cilostazol; low-density lipoprotein receptor; obesity; peroxisome proliferator-activated receptor-γ; proprotein convertase subtilisin/kexin type 9.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Baass A., Dubuc G., Tremblay M., Delvin E.E., O’Loughlin J., Levy E., Davignon J., Lambert M. Plasma PCSK9 is associated with age, sex, and multiple metabolic markers in a population-based sample of children and adolescents. Clin. Chem. 2009;55:1637–1645. doi: 10.1373/clinchem.2009.126987. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
