

Analysis of Dormancy-Associated Transcriptional Networks Reveals a Shared Quiescence Signature in Lung and Colorectal Cancer
- PMID: 36077264
- PMCID: PMC9456317
- DOI: 10.3390/ijms23179869
Analysis of Dormancy-Associated Transcriptional Networks Reveals a Shared Quiescence Signature in Lung and Colorectal Cancer
Abstract
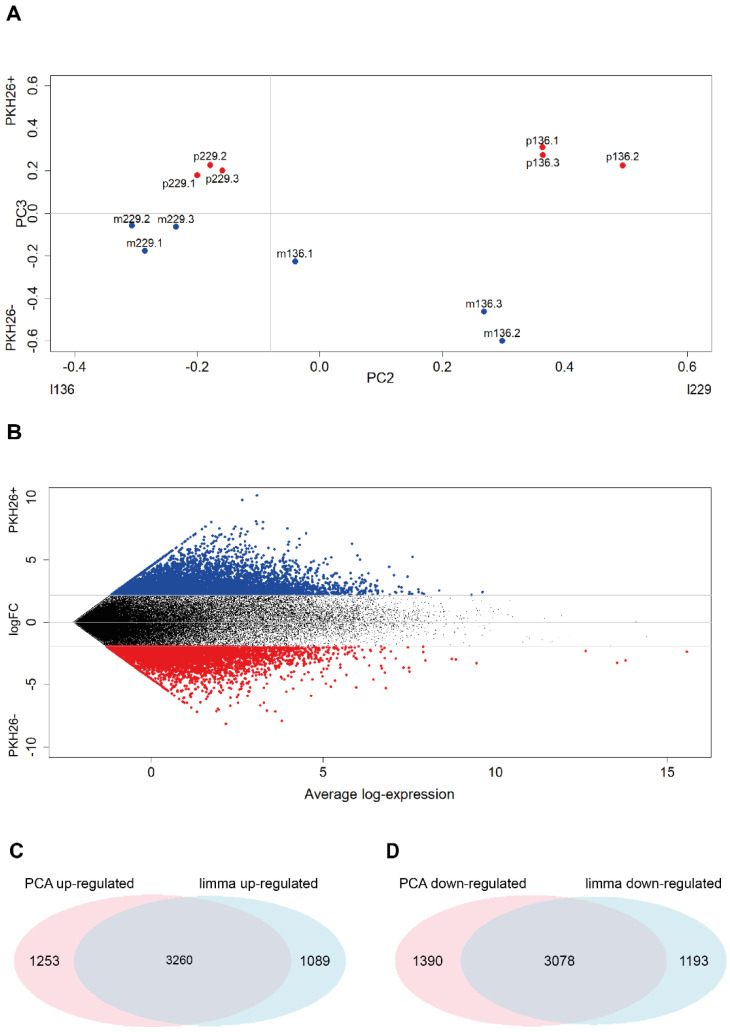
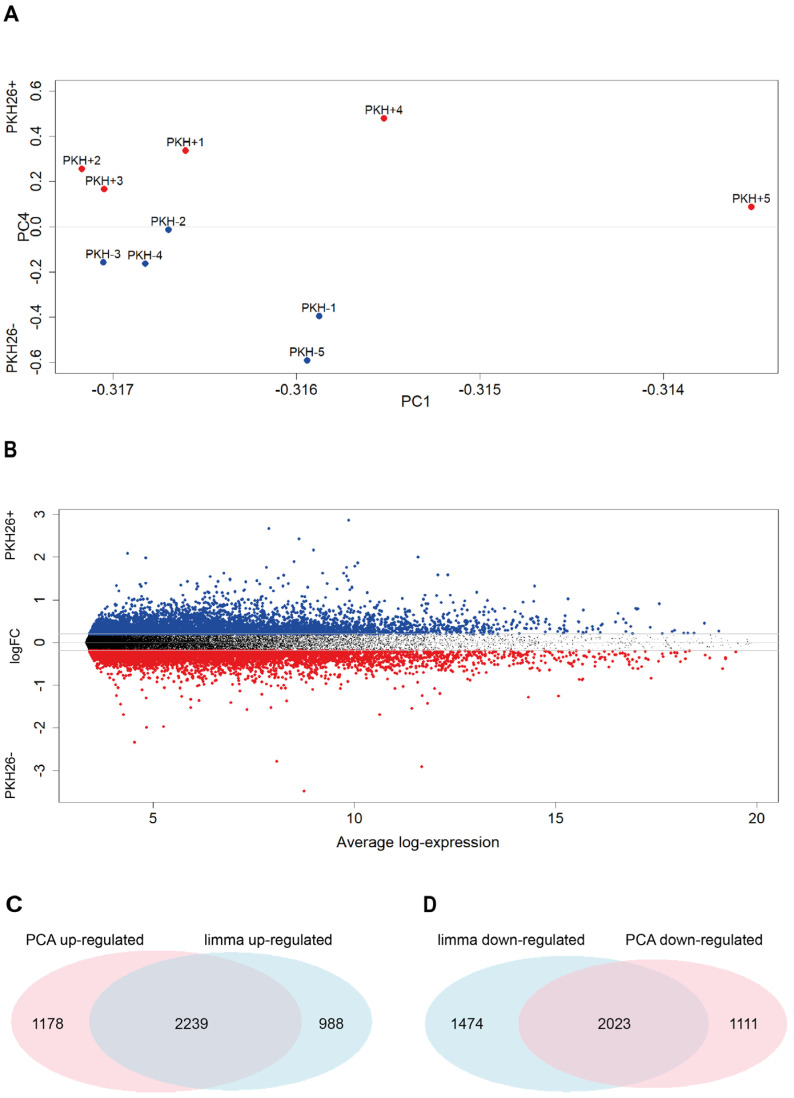
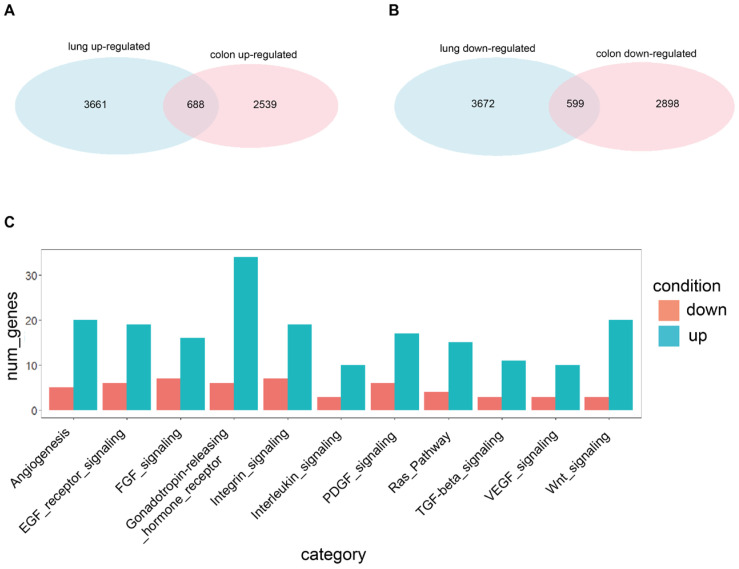
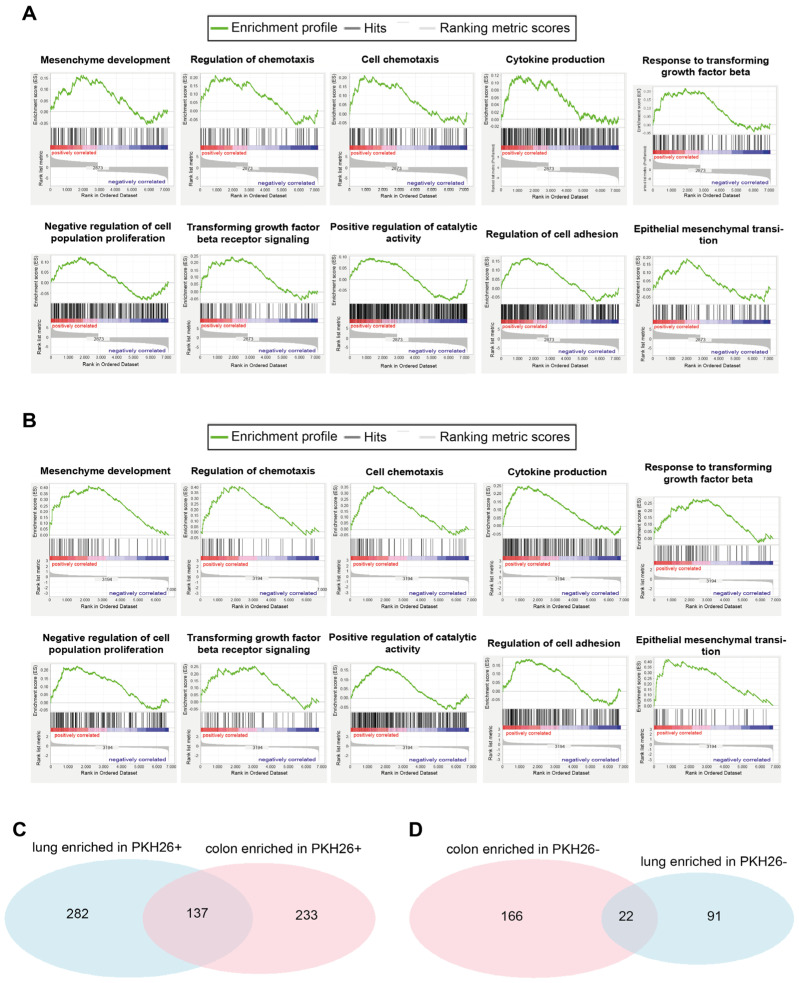
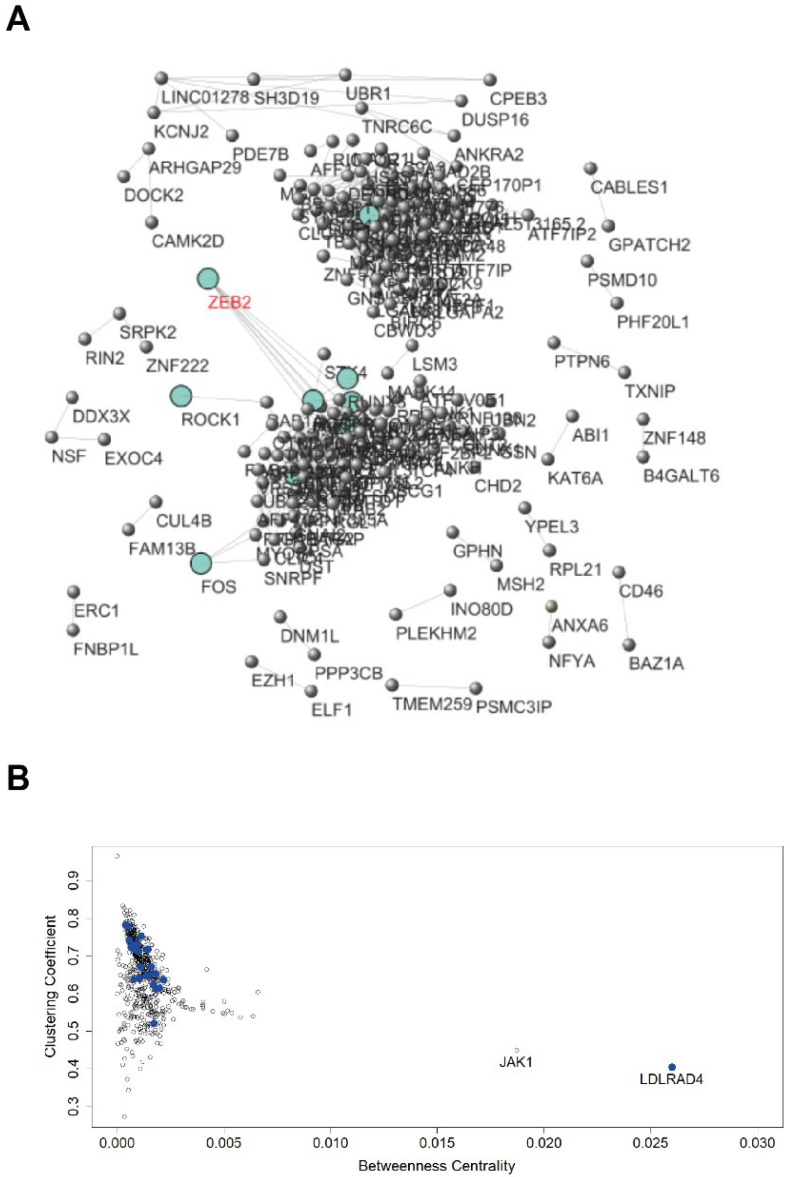
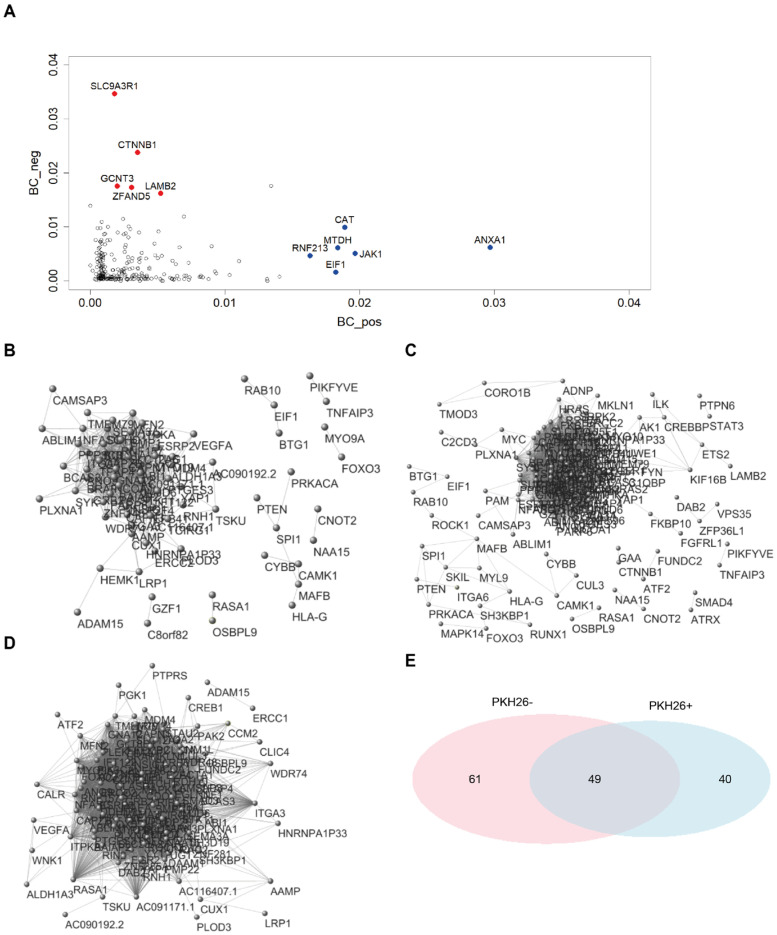
Quiescent cancer cells (QCCs) are a common feature of solid tumors, representing a major obstacle to the long-term success of cancer therapies. We isolated QCCs ex vivo from non-small cell lung cancer (NSCLC) and colorectal cancer (CRC) xenografts with a label-retaining strategy and compared QCCs gene expression profiles to identify a shared "quiescence signature". Principal Component Analysis (PCA) revealed a specific component neatly discriminating quiescent and replicative phenotypes in NSCLC and CRC. The discriminating component showed significant overlapping, with 688 genes in common including ZEB2, a master regulator of stem cell plasticity and epithelial-to-mesenchymal transition (EMT). Gene set enrichment analysis showed that QCCs of both NSCLC and CRC had an increased expression of factors related to stemness/self renewal, EMT, TGF-β, morphogenesis, cell adhesion and chemotaxis, whereas proliferating cells overexpressed Myc targets and factors involved in RNA metabolism. Eventually, we analyzed in depth by means of a complex network approach, both the 'morphogenesis module' and the subset of differentially expressed genes shared by NCSLC and CRC. This allowed us to recognize different gene regulation network wiring for quiescent and proliferating cells and to underpin few genes central for network integration that may represent new therapeutic vulnerabilities. Altogether, our results highlight common regulatory pathways in QCCs of lung and colorectal tumors that may be the target of future therapeutic interventions.
Keywords: colorectal cancer; dormancy; lung cancer; quiescence.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
