

Genomic and phenotypic characterization of 404 individuals with neurodevelopmental disorders caused by CTNNB1 variants
- PMID: 36083290
- PMCID: PMC9939054
- DOI: 10.1016/j.gim.2022.08.006
Genomic and phenotypic characterization of 404 individuals with neurodevelopmental disorders caused by CTNNB1 variants
Abstract
Purpose: Germline loss-of-function variants in CTNNB1 cause neurodevelopmental disorder with spastic diplegia and visual defects (NEDSDV; OMIM 615075) and are the most frequent, recurrent monogenic cause of cerebral palsy (CP). We investigated the range of clinical phenotypes owing to disruptions of CTNNB1 to determine the association between NEDSDV and CP.
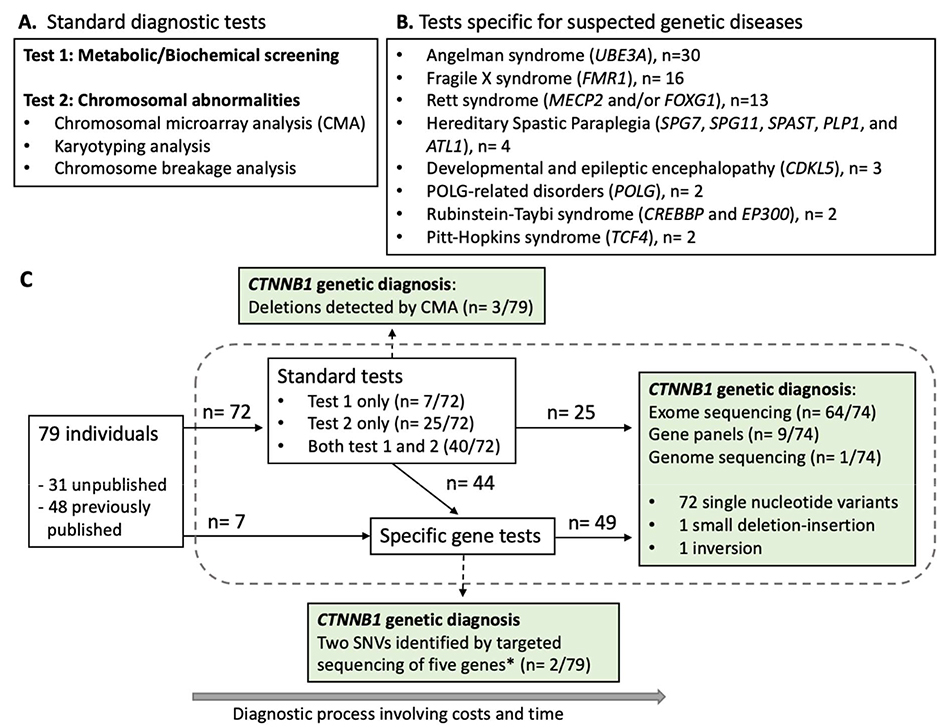
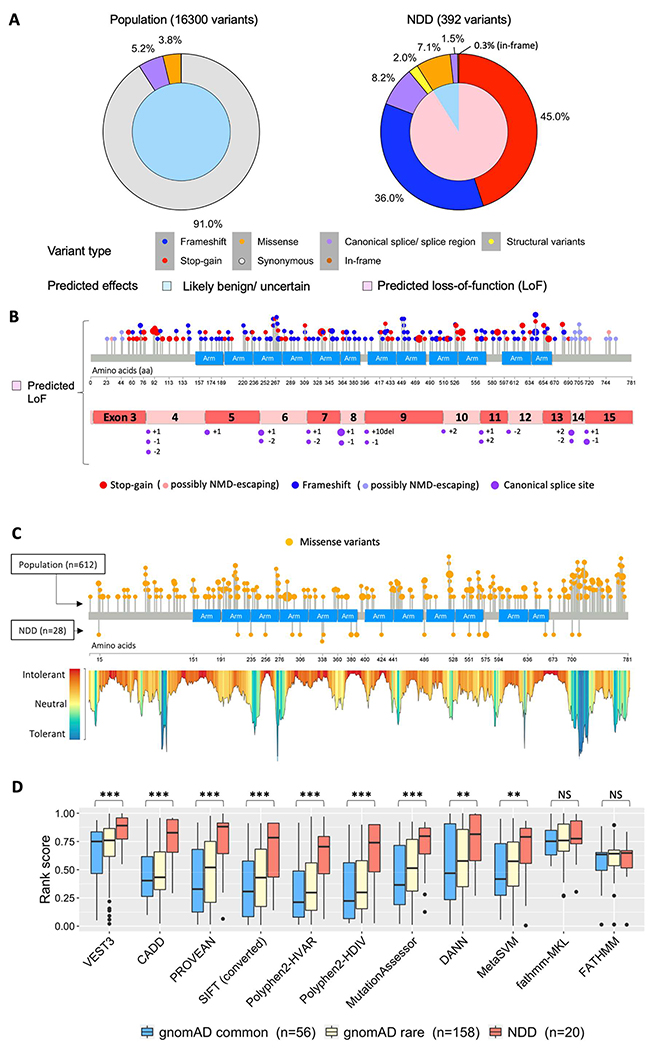
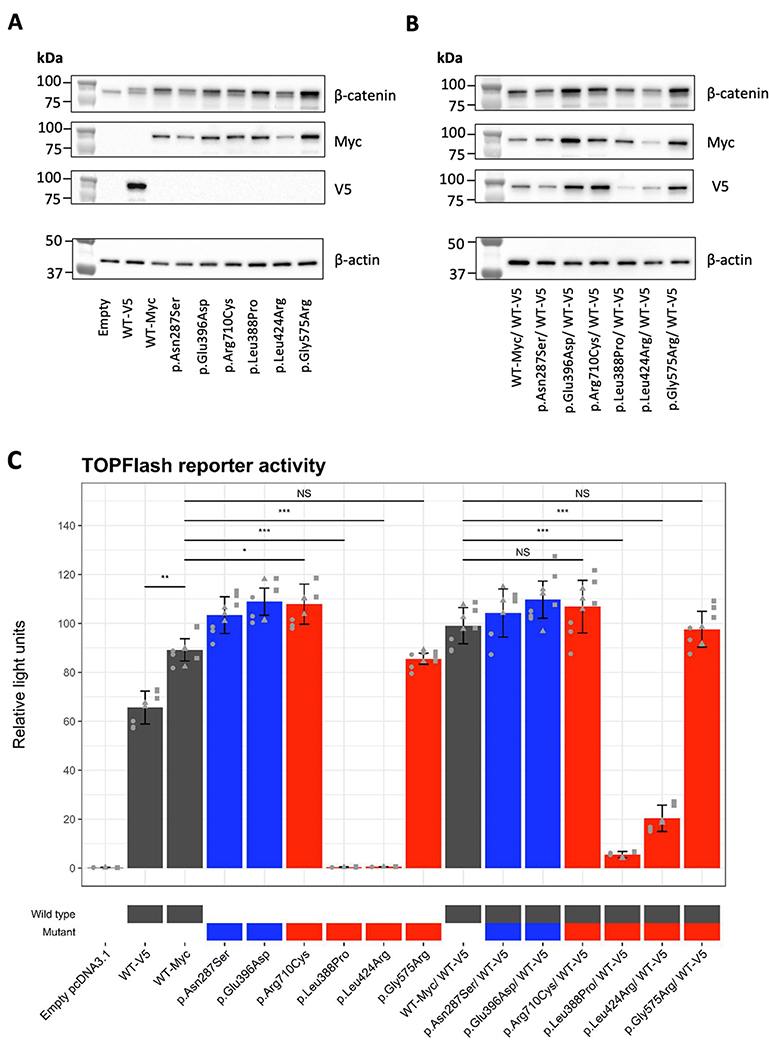
Methods: Genetic information from 404 individuals with collectively 392 pathogenic CTNNB1 variants were ascertained for the study. From these, detailed phenotypes for 52 previously unpublished individuals were collected and combined with 68 previously published individuals with comparable clinical information. The functional effects of selected CTNNB1 missense variants were assessed using TOPFlash assay.
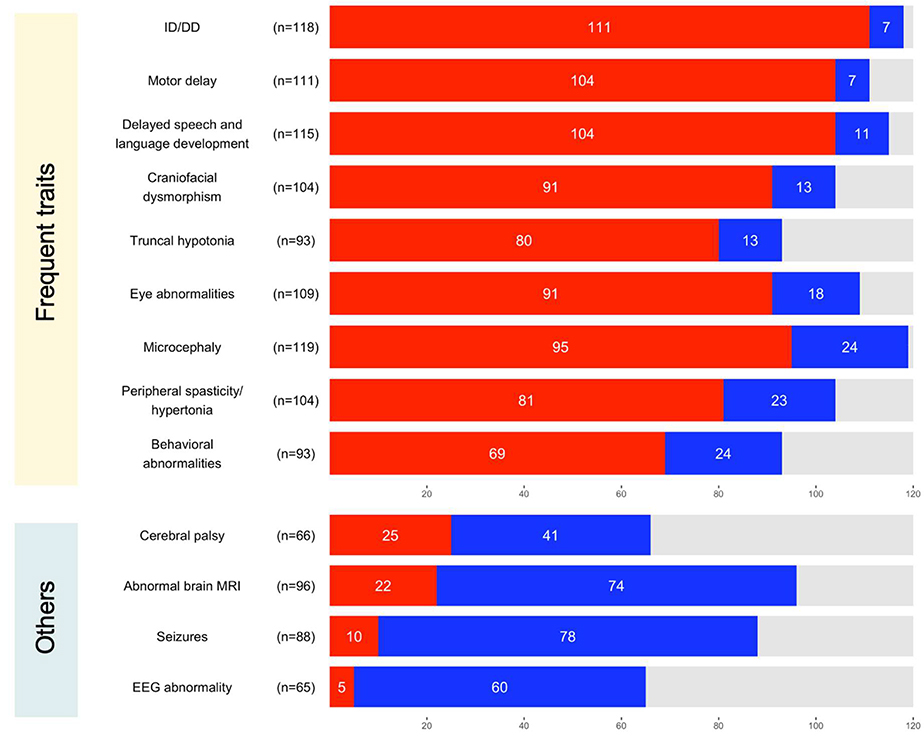
Results: The phenotypes associated with pathogenic CTNNB1 variants were similar. A diagnosis of CP was not significantly associated with any set of traits that defined a specific phenotypic subgroup, indicating that CP is not additional to NEDSDV. Two CTNNB1 missense variants were dominant negative regulators of WNT signaling, highlighting the utility of the TOPFlash assay to functionally assess variants.
Conclusion: NEDSDV is a clinically homogeneous disorder irrespective of initial clinical diagnoses, including CP, or entry points for genetic testing.
Keywords: Autism; Cerebral palsy; Familial exudative vitreoretinopathy; Microcephaly; Wnt beta catenin signaling pathway.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of Interest F.M. and M.M.M. are employees of GeneDX, Inc. All other authors declare no conflict of interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
