

Proteotype coevolution and quantitative diversity across 11 mammalian species
- PMID: 36083897
- PMCID: PMC9462687
- DOI: 10.1126/sciadv.abn0756
Proteotype coevolution and quantitative diversity across 11 mammalian species
Abstract
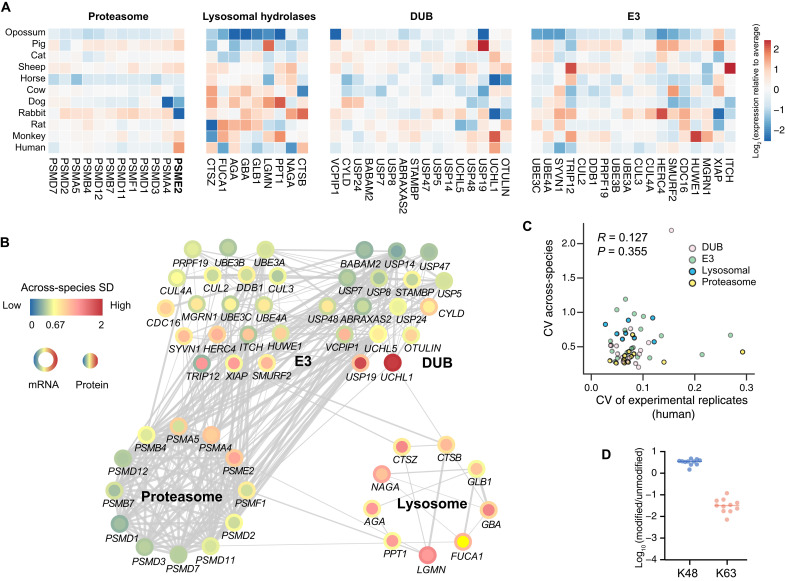
Evolutionary profiling has been largely limited to the nucleotide level. Using consistent proteomic methods, we quantified proteomic and phosphoproteomic layers in fibroblasts from 11 common mammalian species, with transcriptomes as reference. Covariation analysis indicates that transcript and protein expression levels and variabilities across mammals remarkably follow functional role, with extracellular matrix-associated expression being the most variable, demonstrating strong transcriptome-proteome coevolution. The biological variability of gene expression is universal at both interindividual and interspecies scales but to a different extent. RNA metabolic processes particularly show higher interspecies versus interindividual variation. Our results further indicate that while the ubiquitin-proteasome system is strongly conserved in mammals, lysosome-mediated protein degradation exhibits remarkable variation between mammalian lineages. In addition, the phosphosite profiles reveal a phosphorylation coevolution network independent of protein abundance.
Figures




References
-
- Müller J. B., Geyer P. E., Colaço A. R., Treit P. V., Strauss M. T., Oroshi M., Doll S., Winter S. V., Bader J. M., Köhler N., Theis F., Santos A., Mann M., The proteome landscape of the kingdoms of life. Nature 582, 592–596 (2020). - PubMed
-
- Wang Z.-Y., Leushkin E., Liechti A., Ovchinnikova S., Mößinger K., Brüning T., Rummel C., Grützner F., Cardoso-Moreira M., Janich P., Gatfield D., Diagouraga B., de Massy B., Gill M. E., Peters A. H. F. M., Anders S., Kaessmann H., Transcriptome and translatome co-evolution in mammals. Nature 588, 642–647 (2020). - PMC - PubMed
-
- Aebersold R., Mann M., Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355 (2016). - PubMed
-
- Navarro P., Kuharev J., Gillet L. C., Bernhardt O. M., Lean B. M., Röst H. L., Tate S. A., Tsou C.-C., Reiter L., Distler U., Rosenberger G., Perez-Riverol Y., Nesvizhskii A. I., Aebersold R., Tenzer S., A multicenter study benchmarks software tools for label-free proteome quantification. Nat. Biotechnol. 34, 1130–1136 (2016). - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
