

Multidisciplinary team directed analysis of whole genome sequencing reveals pathogenic non-coding variants in molecularly undiagnosed inherited retinal dystrophies
- PMID: 36084042
- PMCID: PMC9896476
- DOI: 10.1093/hmg/ddac227
Multidisciplinary team directed analysis of whole genome sequencing reveals pathogenic non-coding variants in molecularly undiagnosed inherited retinal dystrophies
Abstract
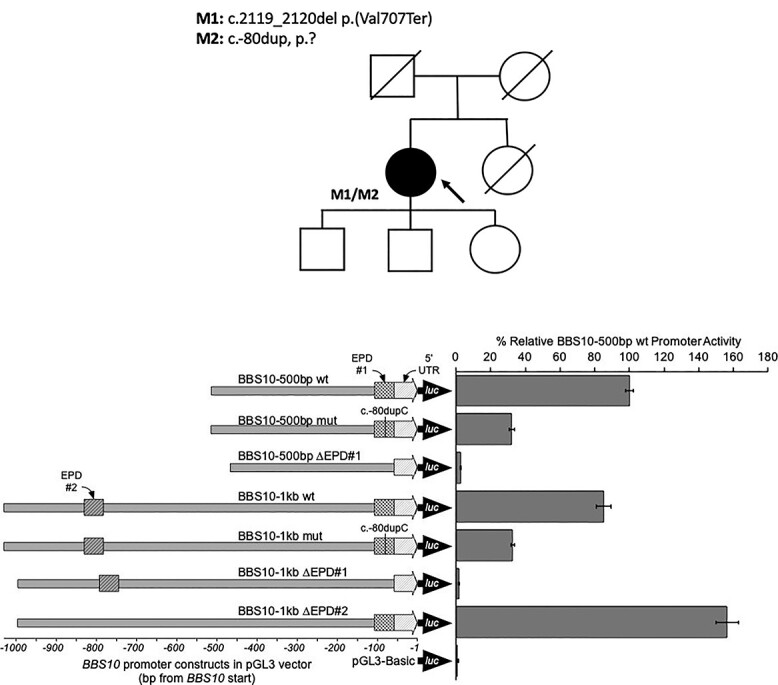
The purpose of this paper is to identify likely pathogenic non-coding variants in inherited retinal dystrophy (IRD) genes, using genome sequencing (GS). Patients with IRD were recruited to the study and underwent comprehensive ophthalmological evaluation and GS. The results of GS were investigated through virtual gene panel analysis, and plausible pathogenic variants and clinical phenotype evaluated by the multidisciplinary team (MDT) discussion. For unsolved patients in whom a specific gene was suspected to harbor a missed pathogenic variant, targeted re-analysis of non-coding regions was performed on GS data. Candidate variants were functionally tested by messenger RNA analysis, minigene or luciferase reporter assays. Previously unreported, likely pathogenic, non-coding variants in 7 genes (PRPF31, NDP, IFT140, CRB1, USH2A, BBS10 and GUCY2D), were identified in 11 patients. These were shown to lead to mis-splicing (PRPF31, IFT140, CRB1 and USH2A) or altered transcription levels (BBS10 and GUCY2D). MDT-led, phenotype-driven, non-coding variant re-analysis of GS is effective in identifying the missing causative alleles.
© The Author(s) 2022. Published by Oxford University Press.
Figures





References
-
- Pontikos, N., Arno, G., Jurkute, N., Schiff, E., Ba-Abbad, R., Malka, S., Gimenez, A., Georgiou, M., Wright, G., Armengo, M.et al. (2020) Genetic basis of inherited retinal disease in a molecularly characterized cohort of more than 3000 families from the United Kingdom. Ophthalmology, 127, 1384–1394. - PMC - PubMed
-
- Tsui, I., Song, B.J., Lin, C.S. and Tsang, S.H. (2018) A practical approach to retinal dystrophies. Adv. Exp. Med. Biol., 1085, 245–259. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
