

Comparative analysis of two genomes of Chlamydia pecorum isolates from an Alpine chamois and a water buffalo
- PMID: 36088280
- PMCID: PMC9464383
- DOI: 10.1186/s12864-022-08860-7
Comparative analysis of two genomes of Chlamydia pecorum isolates from an Alpine chamois and a water buffalo
Abstract
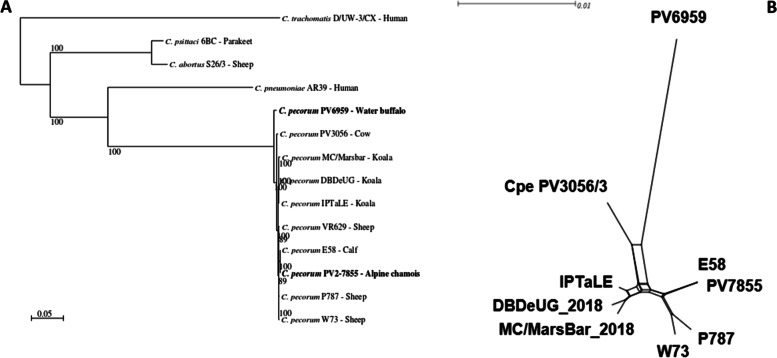
Background: To date, whole genome sequencing has been performed mainly for isolates of Chlamydia trachomatis, C. pneumoniae, C. psittaci and C. abortus, but only a few isolates of C. pecorum have been entirely sequenced and this makes it difficult to understand its diversity and population structure. In this study the genome of two C. pecorum strains isolated from the lung of an Alpine chamois affected with pneumonia (isolate PV7855) and the brain of a water buffalo affected with meningoencephalomyelitis (isolate PV6959), were completely sequenced with MiSeq system (Illumina) and analyzed in their most polymorphic regions.
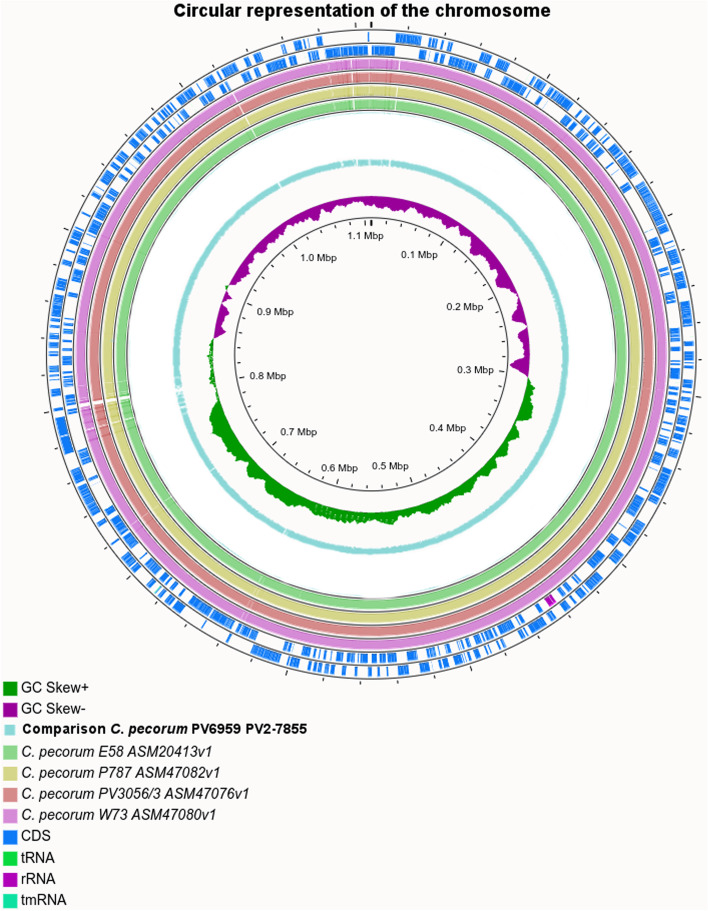
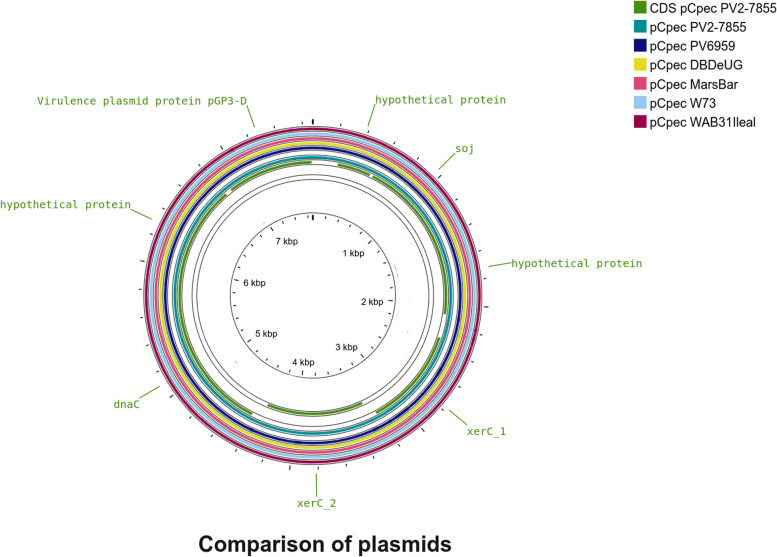
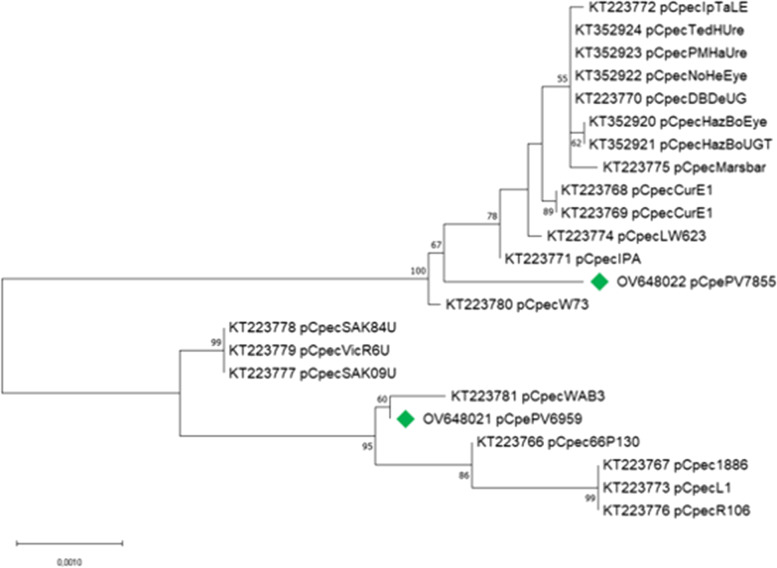
Results: The genome length and GC content of the two isolates were found to be consistent with other C. pecorum isolates and the gene content of polymorphic membrane proteins and plasticity zone was found to be very similar. Some differences were observed in the phospholipase genes for both isolates and in the number of genes in the plasticity zone, such as the presence of some hypothetical proteins in PV6959, not present in any other genomes analyzed in this study. Interestingly, PV6959 possesses an extra pmp and has an incomplete tryptophan biosynthesis operon. Plasmids were detected in both isolates.
Conclusions: Genome sequencing of the two C. pecorum strains did not reveal differences in length and GC content despite the origin from different animal species with different clinical disease. In the plasticity zone, the differences in the genes pattern might be related to the onset of specific symptoms or infection of specific hosts. The absence of a tryptophan biosynthesis pathway in PV6959 may suggest a strict relationship between C. pecorum and its host.
Keywords: Chamois; Chlamydia pecorum; Plasmids; Plasticity zone; Polymorphic membrane protein; Water buffalo; Whole genome sequencing.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
