

Struct2Graph: a graph attention network for structure based predictions of protein-protein interactions
- PMID: 36088285
- PMCID: PMC9464414
- DOI: 10.1186/s12859-022-04910-9
Struct2Graph: a graph attention network for structure based predictions of protein-protein interactions
Abstract
Background: Development of new methods for analysis of protein-protein interactions (PPIs) at molecular and nanometer scales gives insights into intracellular signaling pathways and will improve understanding of protein functions, as well as other nanoscale structures of biological and abiological origins. Recent advances in computational tools, particularly the ones involving modern deep learning algorithms, have been shown to complement experimental approaches for describing and rationalizing PPIs. However, most of the existing works on PPI predictions use protein-sequence information, and thus have difficulties in accounting for the three-dimensional organization of the protein chains.
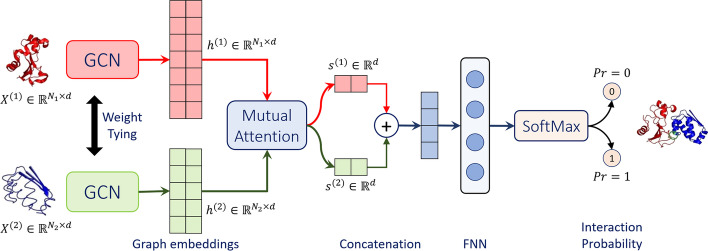
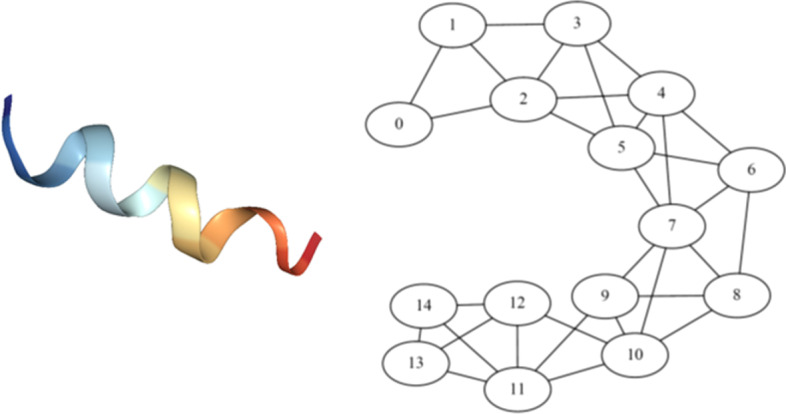
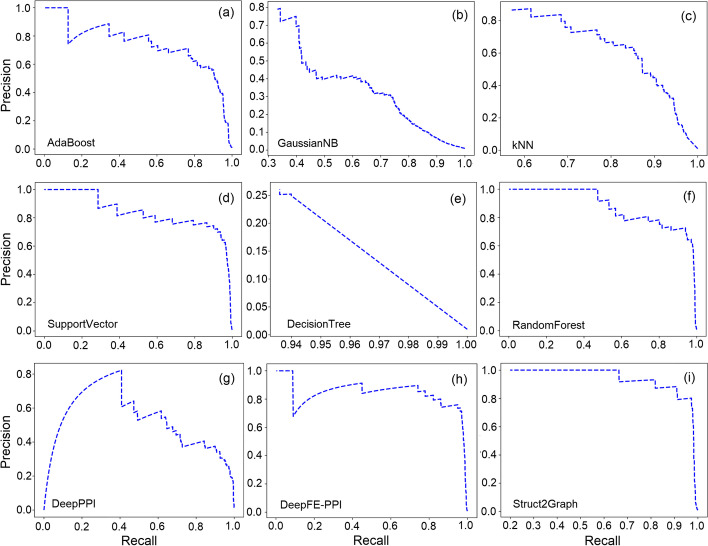
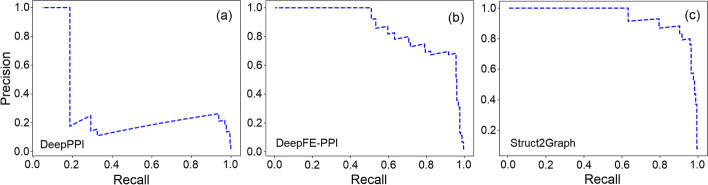
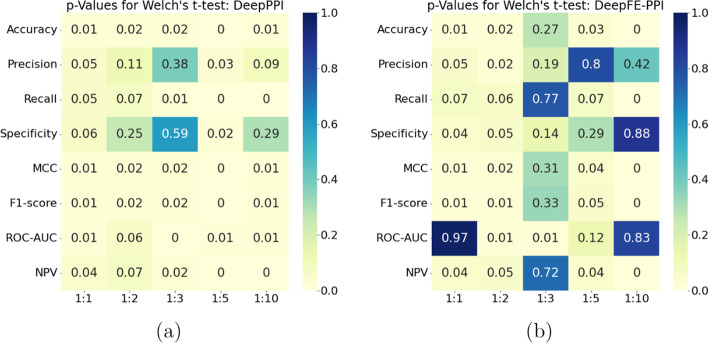
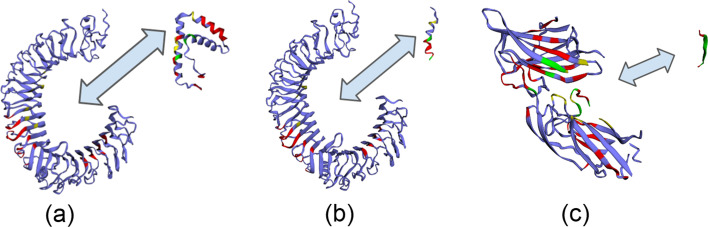
Results: In this study, we address this problem and describe a PPI analysis based on a graph attention network, named Struct2Graph, for identifying PPIs directly from the structural data of folded protein globules. Our method is capable of predicting the PPI with an accuracy of 98.89% on the balanced set consisting of an equal number of positive and negative pairs. On the unbalanced set with the ratio of 1:10 between positive and negative pairs, Struct2Graph achieves a fivefold cross validation average accuracy of 99.42%. Moreover, Struct2Graph can potentially identify residues that likely contribute to the formation of the protein-protein complex. The identification of important residues is tested for two different interaction types: (a) Proteins with multiple ligands competing for the same binding area, (b) Dynamic protein-protein adhesion interaction. Struct2Graph identifies interacting residues with 30% sensitivity, 89% specificity, and 87% accuracy.
Conclusions: In this manuscript, we address the problem of prediction of PPIs using a first of its kind, 3D-structure-based graph attention network (code available at https://github.com/baranwa2/Struct2Graph ). Furthermore, the novel mutual attention mechanism provides insights into likely interaction sites through its unsupervised knowledge selection process. This study demonstrates that a relatively low-dimensional feature embedding learned from graph structures of individual proteins outperforms other modern machine learning classifiers based on global protein features. In addition, through the analysis of single amino acid variations, the attention mechanism shows preference for disease-causing residue variations over benign polymorphisms, demonstrating that it is not limited to interface residues.
Keywords: Deep learning; Graph attention network; Protein–protein interaction; Structure-based prediction.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
