

Loss-of-function variants in the KCNQ5 gene are implicated in genetic generalized epilepsies
- PMID: 36088682
- PMCID: PMC9471468
- DOI: 10.1016/j.ebiom.2022.104244
Loss-of-function variants in the KCNQ5 gene are implicated in genetic generalized epilepsies
Abstract
Background: De novo missense variants in KCNQ5, encoding the voltage-gated K+ channel KV7.5, have been described to cause developmental and epileptic encephalopathy (DEE) or intellectual disability (ID). We set out to identify disease-related KCNQ5 variants in genetic generalized epilepsy (GGE) and their underlying mechanisms.
Methods: 1292 families with GGE were studied by next-generation sequencing. Whole-cell patch-clamp recordings, biotinylation and phospholipid overlay assays were performed in mammalian cells combined with homology modelling.
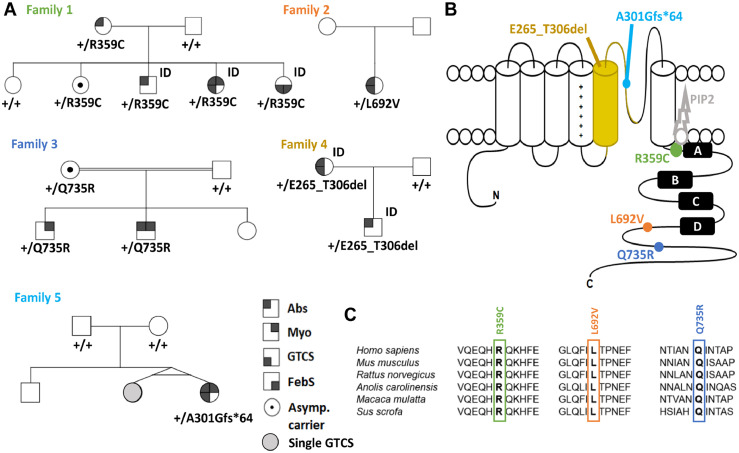
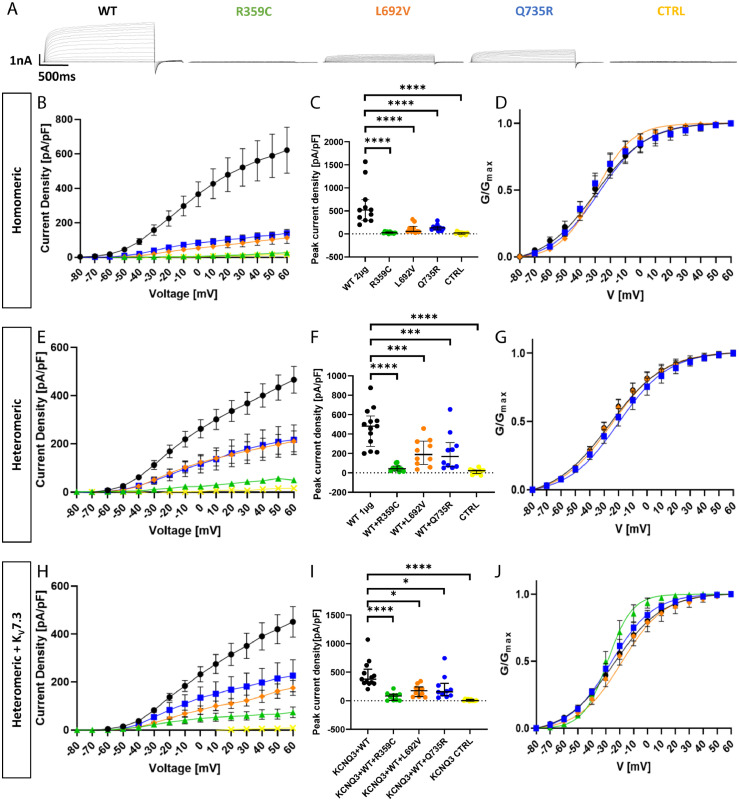
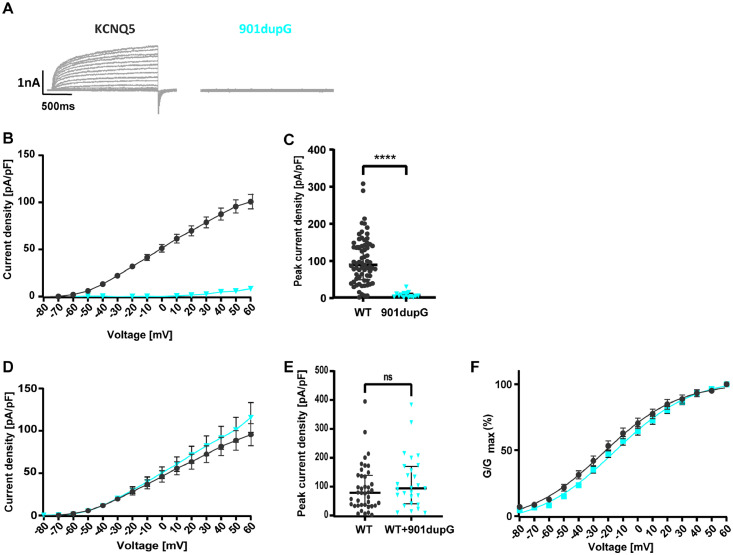
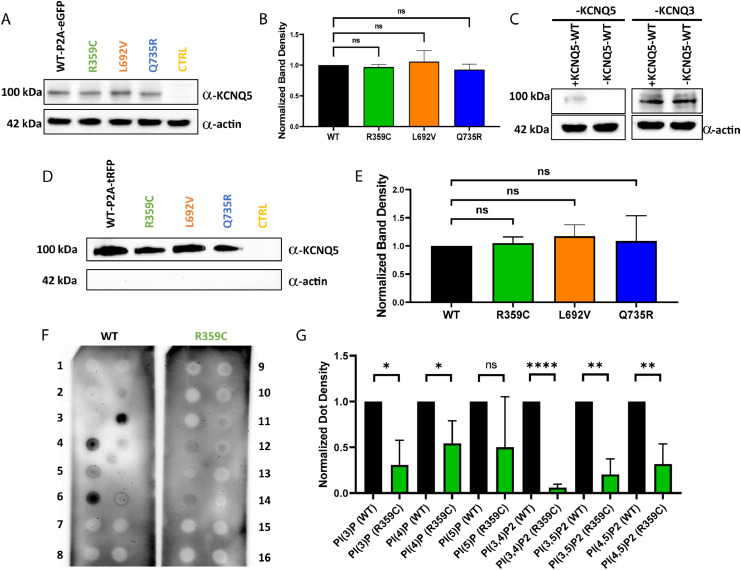
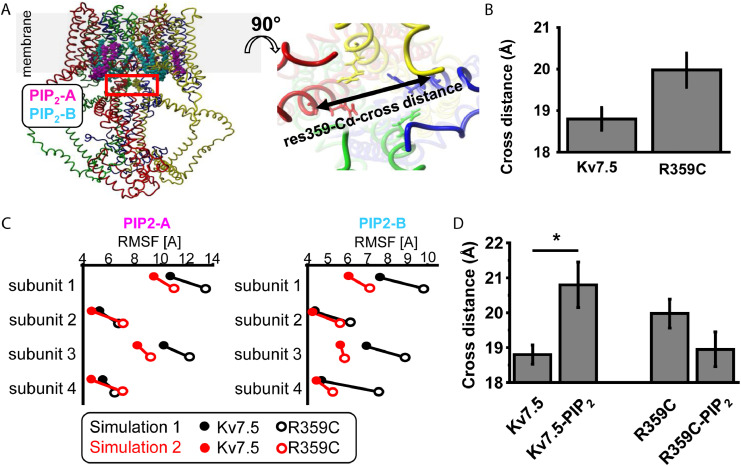
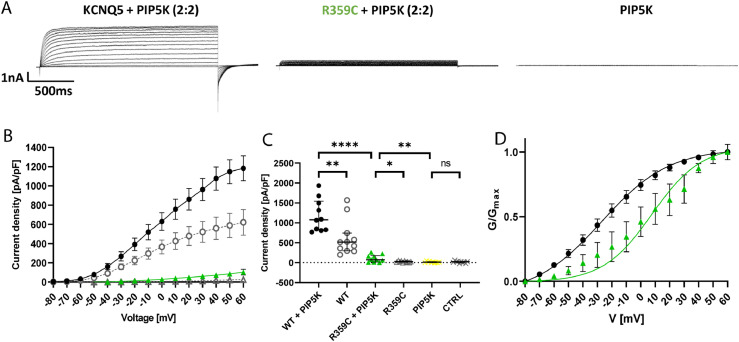
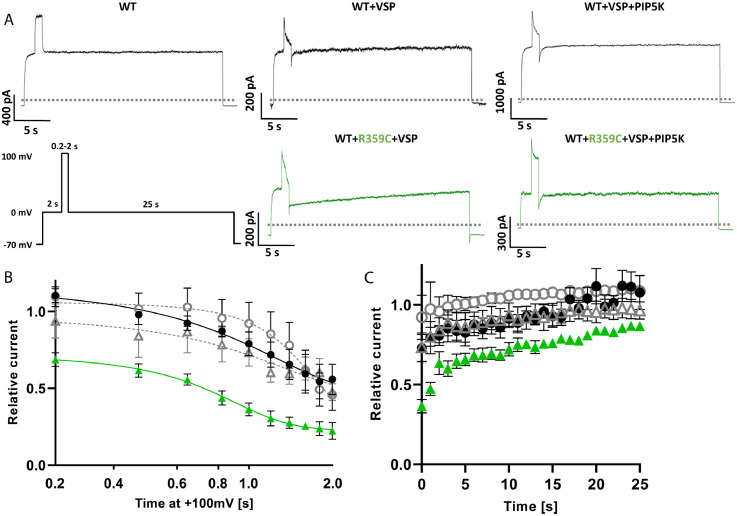
Findings: We identified three deleterious heterozygous missense variants, one truncation and one splice site alteration in five independent families with GGE with predominant absence seizures; two variants were also associated with mild to moderate ID. All missense variants displayed a strongly decreased current density indicating a loss-of-function (LOF). When mutant channels were co-expressed with wild-type (WT) KV7.5 or KV7.5 and KV7.3 channels, three variants also revealed a significant dominant-negative effect on WT channels. Other gating parameters were unchanged. Biotinylation assays indicated a normal surface expression of the variants. The R359C variant altered PI(4,5)P2-interaction.
Interpretation: Our study identified deleterious KCNQ5 variants in GGE, partially combined with mild to moderate ID. The disease mechanism is a LOF partially with dominant-negative effects through functional deficits. LOF of KV7.5 channels will reduce the M-current, likely resulting in increased excitability of KV7.5-expressing neurons. Further studies on network level are necessary to understand which circuits are affected and how this induces generalized seizures.
Funding: DFG/FNR Research Unit FOR-2715 (Germany/Luxemburg), BMBF rare disease network Treat-ION (Germany), foundation 'no epilep' (Germany).
Keywords: Exome sequencing; Genetic generalized epilepsy; KCNQ5; Loss-of-function; Patch-clamp.
Copyright © 2022 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests J. Krüger was financed by a grant from the Deutsche Forschungsgemeinschaft/German Research Foundation (DFG), during the conduct of the study; Dr. Schubert has nothing to disclose; Dr. Kegele has nothing to disclose; A. Labalme has nothing to disclose; Dr. Mao has nothing to disclose; J. Heighway has nothing to disclose; Dr. Seebohm has nothing to disclose; Dr. Yan has nothing to disclose; M. Koko reports grants from DAAD, outside the submitted work; Dr. Aslan has nothing to disclose; Dr. Caglayan has nothing to disclose; Dr. Steinhoff has nothing to disclose; Dr. Weber has nothing to disclose; Dr. Keo Kosal has nothing to disclose; Dr. Berkovic reports grants from NHMRC, during the conduct of the study; grants from UCB Pharma, grants from Eisai, grants from SciGen, personal fees from Bionomics, personal fees from Athena Diagnostics, outside the submitted work; In addition, Dr. Berkovic has a patent Methods of treatment, and diagnosis of epilepsy by detecting mutations in the SCN1A gene with royalties paid to Patent held by Bionomics Inc. Licensed to Athena Diagnostics; Genetics Technologies Ltd, a patent Diagnostic and Therapeutic Methods for EFMR (Epilepsy and Mental Retardation Limited to Females) with royalties paid to Licensed to Athena Diagnostics, and a patent A gene and mutations thereof associated with seizure and movement disorders (PRRT2) with royalties paid to Licensed to Athena Diagnostics; Dr. Hildebrand has nothing to disclose; Dr. Petrou reports personal fees and other from Praxis Precision Medicines, outside the submitted work; and Dr. Petrou works for a company, Praxis Precision Medicines that develop therapies for neurogenetic disorders such as KCNQ5 (but this is not currently under any consideration); Drs. Krause and May has report grants from the Fond Nationale de la Recherche in Luxembourg; Dr. Lesca has nothing to disclose; Dr. Maljevic has nothing to disclose; Dr. Lerche reports grants from the German Research Foundation (DFG), from the Federal Ministry for Education and Research (BMBF), grants from Foundation no epilep, during the conduct of the study; outside the submitted work, Dr. Lerche reports a grant from the Else-Kröner Fresenius Foundation (EKFS), a grant and personal fees from Bial, a grant from Boehringer Ingelheim, personal fees from Eisai, personal fees from UCB/Zogenix, personal fees from Arvelle/Angelini Pharma, personal fees from Desitin, and personal fees from IntraBio.
Figures
References
-
- Lerche C, Scherer CR, Seebohm G, et al. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem. 2000;275(29):22395–22400. - PubMed
-
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275(31):24089–24095. - PubMed
-
- Wang H. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science. 1998;282(5395):1890–1893. - PubMed
-
- Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12(1):17–23. - PubMed
-
- Biervert C, Schroeder BC, Kubisch C, et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279(5349):403–406. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
