

Hunting for the cause: Evidence for prion-like mechanisms in Huntington's disease
- PMID: 36090278
- PMCID: PMC9448931
- DOI: 10.3389/fnins.2022.946822
Hunting for the cause: Evidence for prion-like mechanisms in Huntington's disease
Abstract
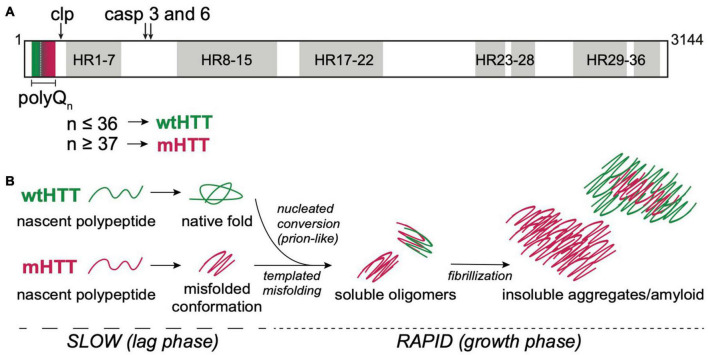
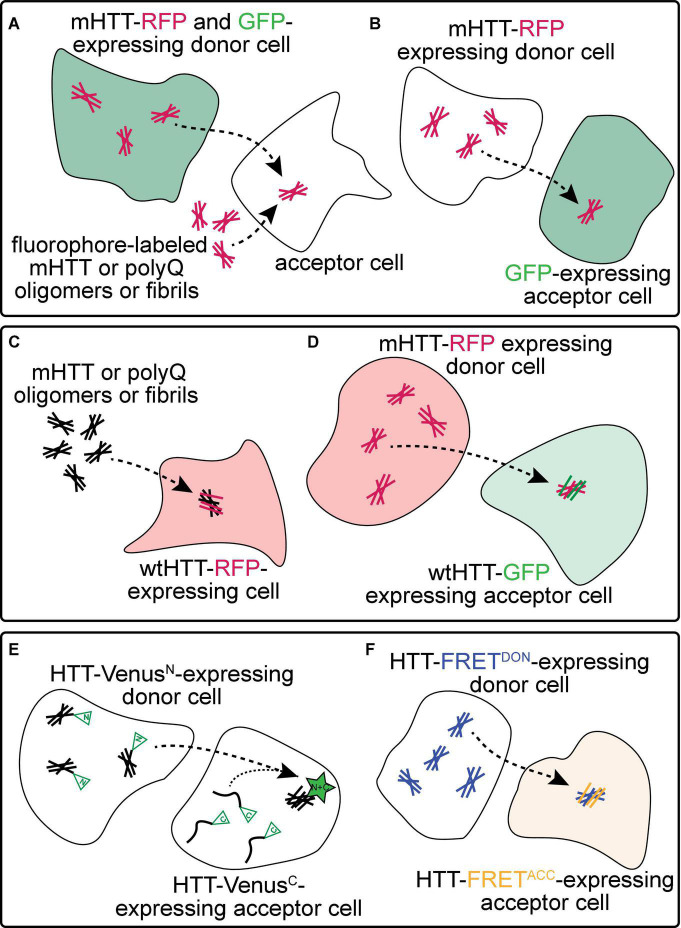
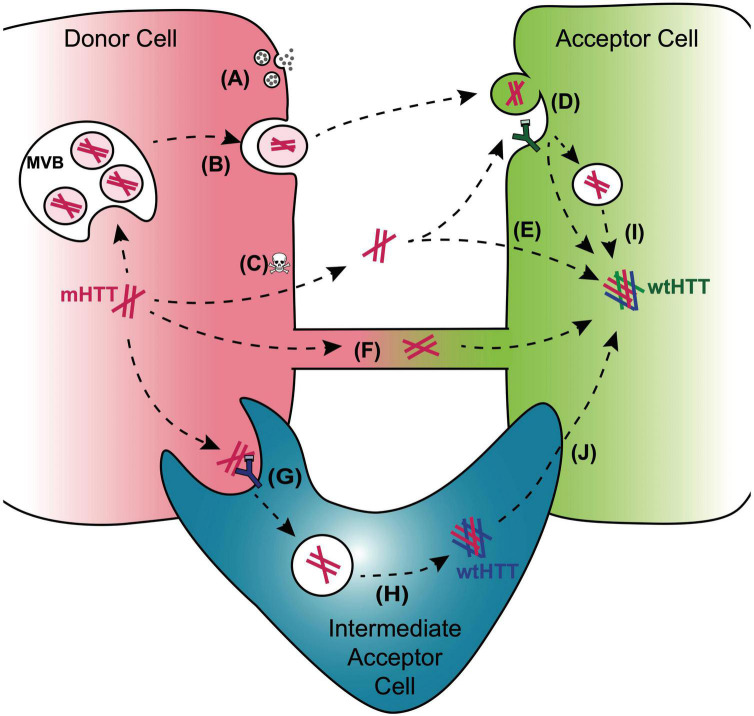
The hypothesis that pathogenic protein aggregates associated with neurodegenerative diseases spread from cell-to-cell in the brain in a manner akin to infectious prions has gained substantial momentum due to an explosion of research in the past 10-15 years. Here, we review current evidence supporting the existence of prion-like mechanisms in Huntington's disease (HD), an autosomal dominant neurodegenerative disease caused by expansion of a CAG repeat tract in exon 1 of the huntingtin (HTT) gene. We summarize information gained from human studies and in vivo and in vitro models of HD that strongly support prion-like features of the mutant HTT (mHTT) protein, including potential involvement of molecular features of mHTT seeds, synaptic structures and connectivity, endocytic and exocytic mechanisms, tunneling nanotubes, and nonneuronal cells in mHTT propagation in the brain. We discuss mechanisms by which mHTT aggregate spreading and neurotoxicity could be causally linked and the potential benefits of targeting prion-like mechanisms in the search for new disease-modifying therapies for HD and other fatal neurodegenerative diseases.
Keywords: Huntington’s disease; aggregate seed; aggregate spread; mutant huntingtin; polyglutamine; prion-like transmission; protein aggregate.
Copyright © 2022 Donnelly, Coleman, Fuller, Reed, Smerina, Tomlinson and Pearce.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous