

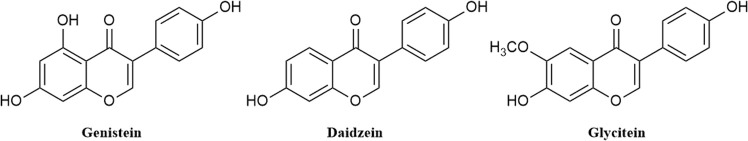
Discovery of genistein derivatives as potential SARS-CoV-2 main protease inhibitors by virtual screening, molecular dynamics simulations and ADMET analysis
- PMID: 36091808
- PMCID: PMC9452787
- DOI: 10.3389/fphar.2022.961154
Discovery of genistein derivatives as potential SARS-CoV-2 main protease inhibitors by virtual screening, molecular dynamics simulations and ADMET analysis
Abstract
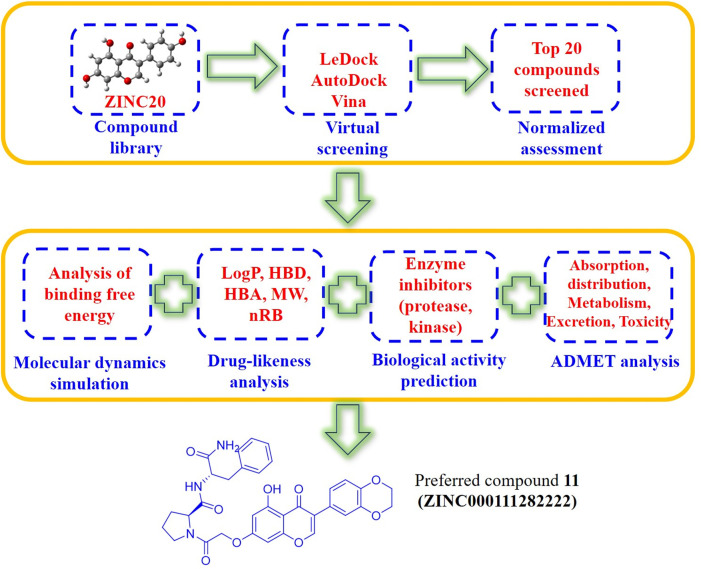
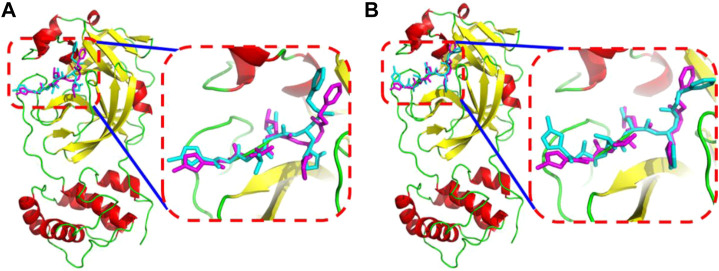
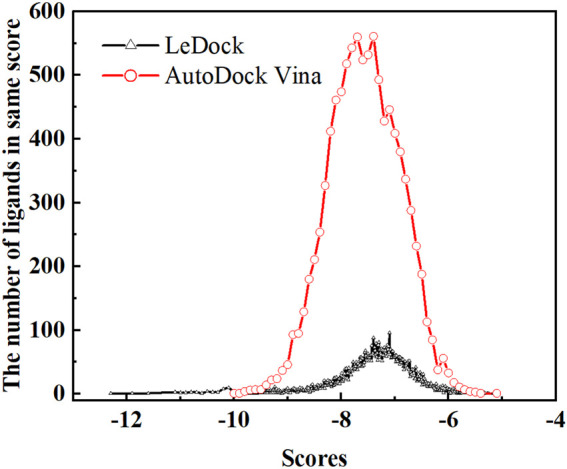
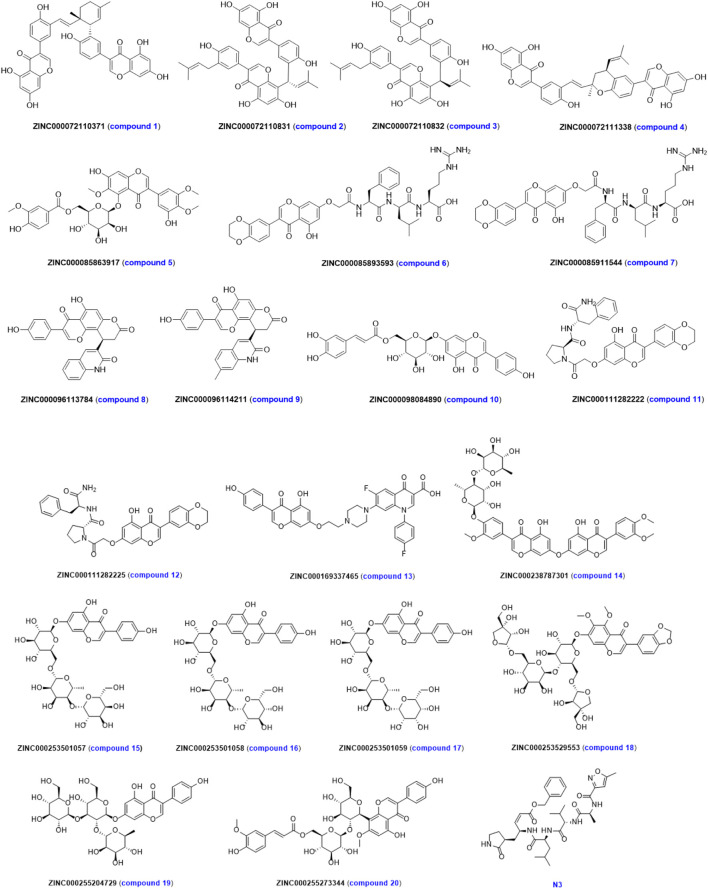
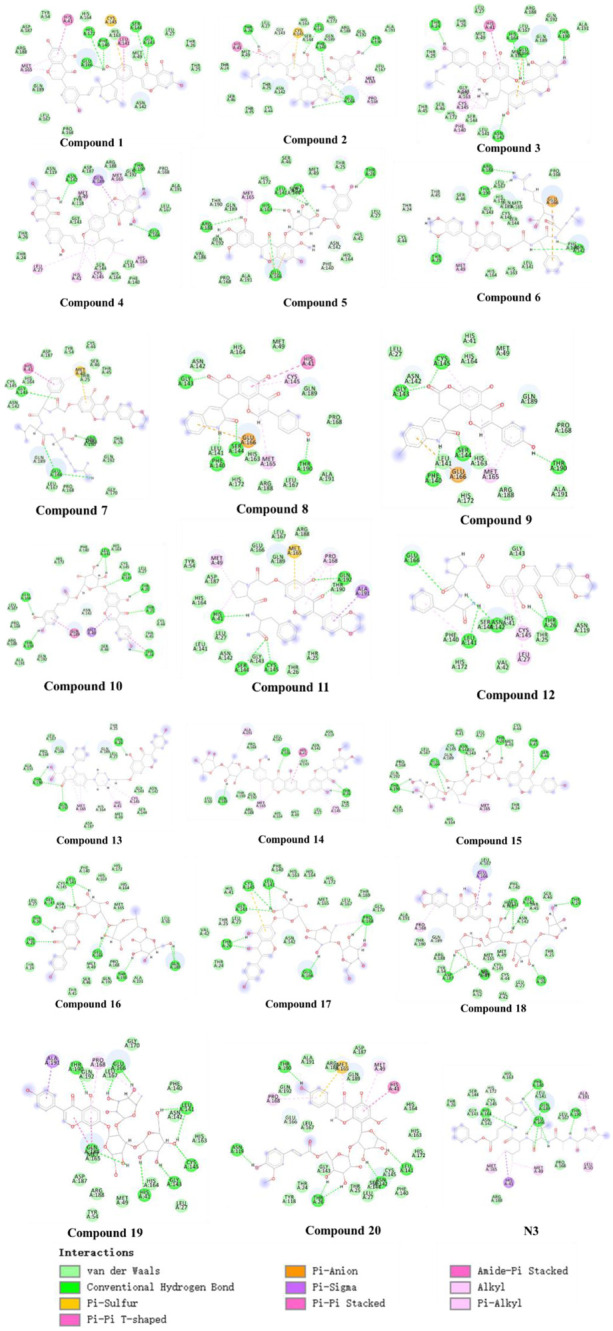
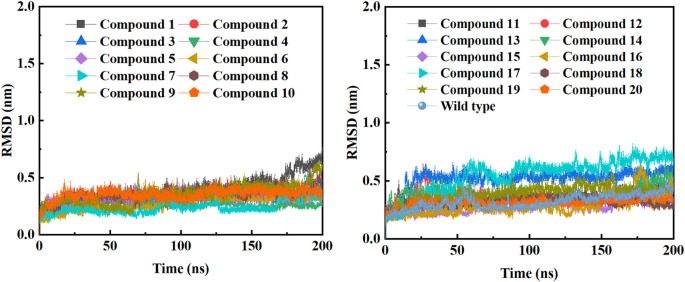
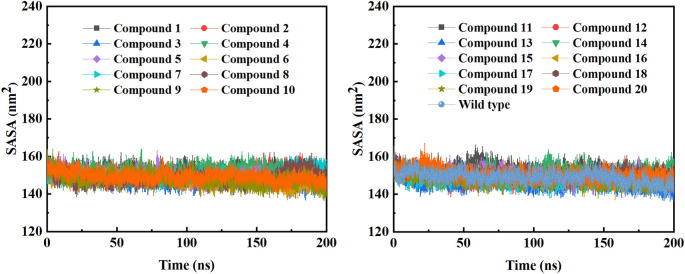
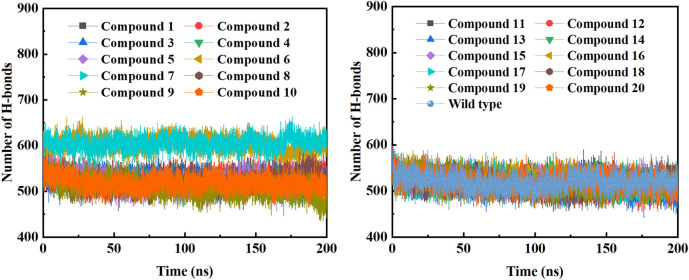
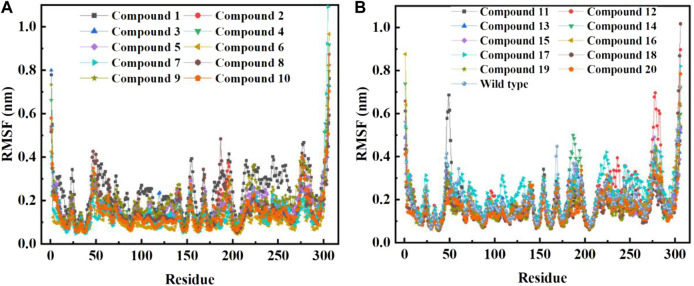
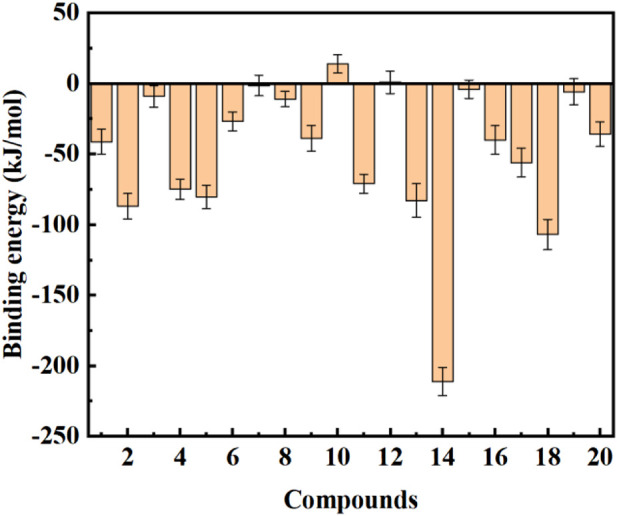
Background: Due to the constant mutation of virus and the lack of specific therapeutic drugs, the coronavirus disease 2019 (COVID-19) pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) still poses a huge threat to the health of people, especially those with underlying diseases. Therefore, drug discovery against the SARS-CoV-2 remains of great significance. Methods: With the main protease of virus as the inhibitor target, 9,614 genistein derivatives were virtually screened by LeDock and AutoDock Vina, and the top 20 compounds with highest normalized scores were obtained. Molecular dynamics simulations were carried out for studying interactions between these 20 compounds and the target protein. The drug-like properties, activity, and ADMET of these compounds were also evaluated by DruLiTo software or online server. Results: Twenty compounds, including compound 11, were screened by normalized molecular docking, which could bind to the target through multiple non-bonding interactions. Molecular dynamics simulation results showed that compounds 2, 4, 5, 11, 13, 14, 17, and 18 had the best binding force with the target protein of SARS-CoV-2, and the absolute values of binding free energies all exceeded 50 kJ/mol. The drug-likeness properties indicated that a variety of compounds including compound 11 were worthy of further study. The results of bioactivity score prediction found that compounds 11 and 12 had high inhibitory activities against protease, which indicated that these two compounds had the potential to be further developed as COVID-19 inhibitors. Finally, compound 11 showed excellent predictive ADMET properties including high absorption and low toxicity. Conclusion: These in silico work results show that the preferred compound 11 (ZINC000111282222), which exhibited strong binding to SARS-CoV-2 main protease, acceptable drug-like properties, protease inhibitory activity and ADMET properties, has great promise for further research as a potential therapeutic agent against COVID-19.
Keywords: ADMET; SARS-CoV-2; genistein; molecular docking; molecular dynamics simulation.
Copyright © 2022 Liu, Zhang, Gao, Zhang, Liu, Yang, Liu, Liu and Cheng.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
In silico validation of coumarin derivatives as potential inhibitors against Main Protease, NSP10/NSP16-Methyltransferase, Phosphatase and Endoribonuclease of SARS CoV-2.J Biomol Struct Dyn. 2021 Nov;39(18):7306-7321. doi: 10.1080/07391102.2020.1808075. Epub 2020 Aug 24. J Biomol Struct Dyn. 2021. PMID: 32835632 Free PMC article.
-
Virtual screening, ADMET prediction and dynamics simulation of potential compounds targeting the main protease of SARS-CoV-2.J Biomol Struct Dyn. 2021 Oct;39(17):6617-6632. doi: 10.1080/07391102.2020.1796812. Epub 2020 Jul 25. J Biomol Struct Dyn. 2021. PMID: 32715956 Free PMC article.
-
Computational selection of flavonoid compounds as inhibitors against SARS-CoV-2 main protease, RNA-dependent RNA polymerase and spike proteins: A molecular docking study.Saudi J Biol Sci. 2021 Jan;28(1):448-458. doi: 10.1016/j.sjbs.2020.10.028. Epub 2020 Oct 22. Saudi J Biol Sci. 2021. PMID: 33110386 Free PMC article.
-
Recent advances in chemometric modelling of inhibitors against SARS-CoV-2.Heliyon. 2024 Jan 9;10(2):e24209. doi: 10.1016/j.heliyon.2024.e24209. eCollection 2024 Jan 30. Heliyon. 2024. PMID: 38293468 Free PMC article. Review.
-
In Silico Models for Anti-COVID-19 Drug Discovery: A Systematic Review.Adv Pharmacol Pharm Sci. 2023 Jun 15;2023:4562974. doi: 10.1155/2023/4562974. eCollection 2023. Adv Pharmacol Pharm Sci. 2023. PMID: 37362912 Free PMC article. Review.
Cited by
-
Exploring the Potential Mechanisms of Guanxinshutong Capsules in Treating Pathological Cardiac Hypertrophy based on Network Pharmacology, Computer-Aided Drug Design, and Animal Experiments.ACS Omega. 2024 Apr 10;9(16):18083-18098. doi: 10.1021/acsomega.3c10009. eCollection 2024 Apr 23. ACS Omega. 2024. PMID: 38680308 Free PMC article.
-
Selective inhibitors targeting Fis1/Mid51 protein-protein interactions protect against hypoxia-induced damage in cardiomyocytes.Front Pharmacol. 2023 Dec 21;14:1275370. doi: 10.3389/fphar.2023.1275370. eCollection 2023. Front Pharmacol. 2023. PMID: 38192411 Free PMC article.
-
Identification, screening, and comprehensive evaluation of novel thrombin inhibitory peptides from the hirudo produced using pepsin.Front Pharmacol. 2024 Nov 21;15:1460053. doi: 10.3389/fphar.2024.1460053. eCollection 2024. Front Pharmacol. 2024. PMID: 39640485 Free PMC article.
-
The influence of COVID-19 on colorectal cancer was investigated using bioinformatics and systems biology techniques.Front Med (Lausanne). 2023 Jun 30;10:1169562. doi: 10.3389/fmed.2023.1169562. eCollection 2023. Front Med (Lausanne). 2023. PMID: 37457582 Free PMC article.
-
Targeting Human Proteins for Antiviral Drug Discovery and Repurposing Efforts: A Focus on Protein Kinases.Viruses. 2023 Feb 19;15(2):568. doi: 10.3390/v15020568. Viruses. 2023. PMID: 36851782 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Miscellaneous