

Comparative Genomics Provides Etiologic and Biological Insight into Melanoma Subtypes
- PMID: 36098958
- PMCID: PMC9716259
- DOI: 10.1158/2159-8290.CD-22-0603
Comparative Genomics Provides Etiologic and Biological Insight into Melanoma Subtypes
Abstract
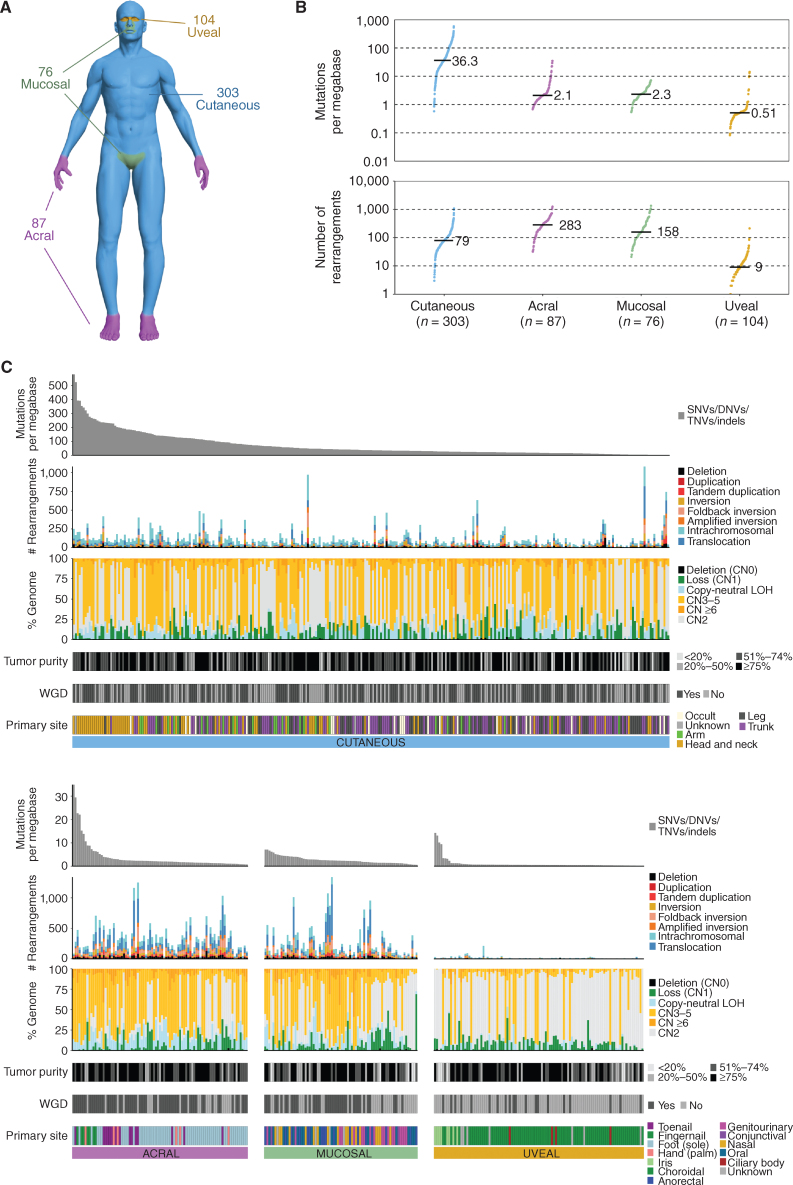
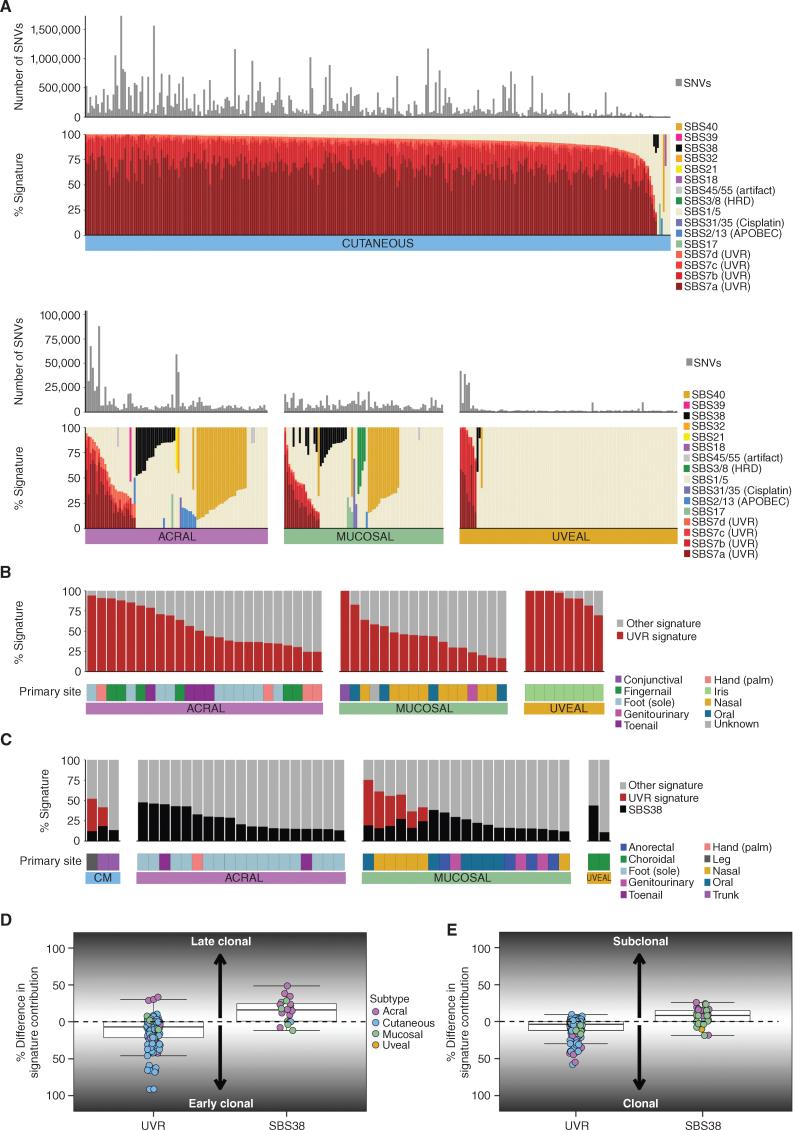
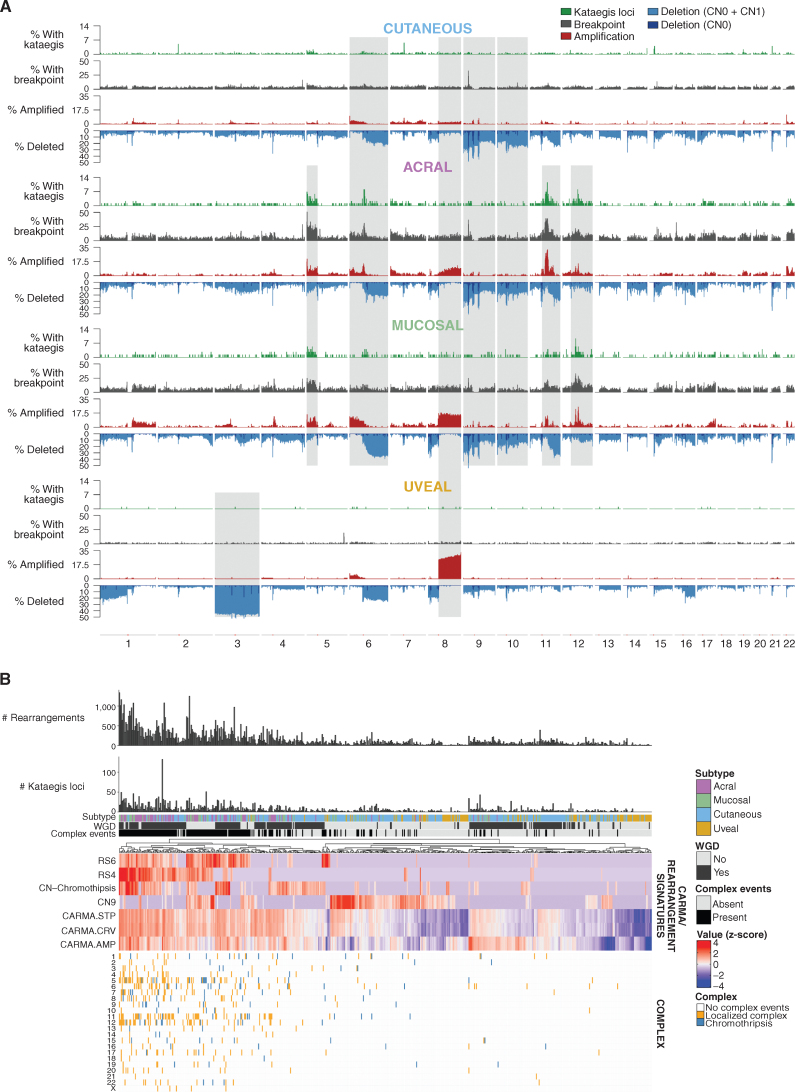
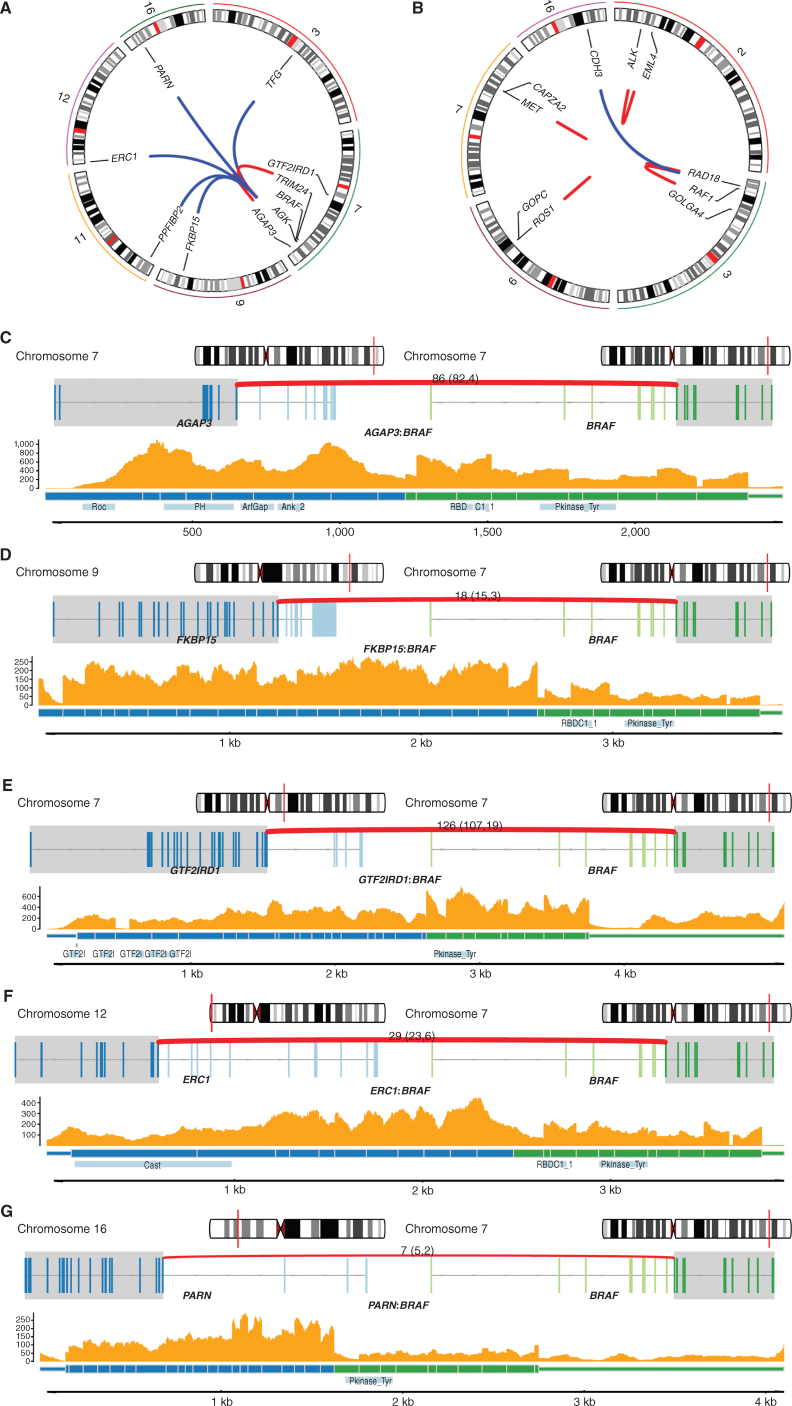
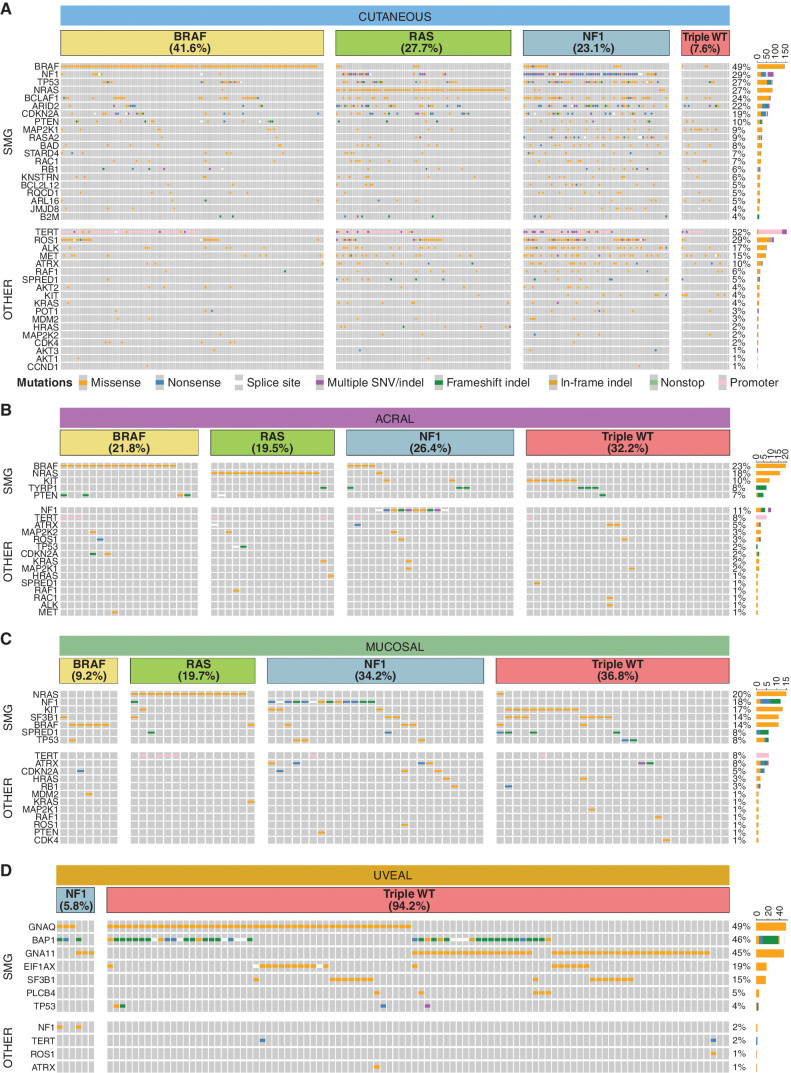
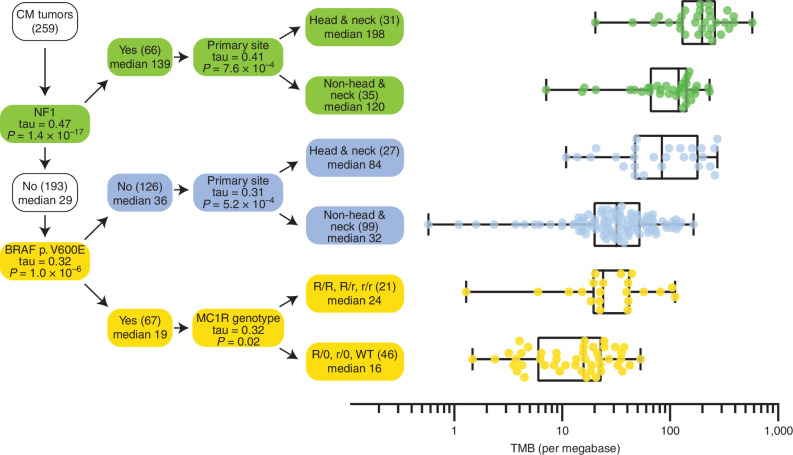
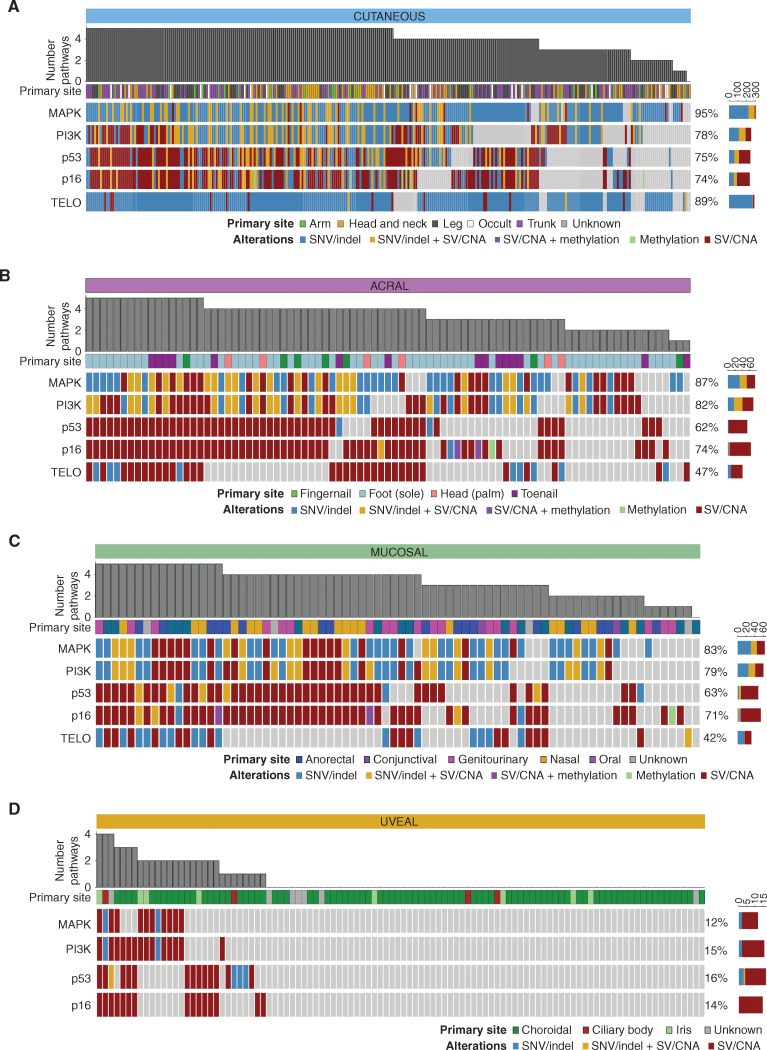
Melanoma is a cancer of melanocytes, with multiple subtypes based on body site location. Cutaneous melanoma is associated with skin exposed to ultraviolet radiation; uveal melanoma occurs in the eyes; mucosal melanoma occurs in internal mucous membranes; and acral melanoma occurs on the palms, soles, and nail beds. Here, we present the largest whole-genome sequencing study of melanoma to date, with 570 tumors profiled, as well as methylation and RNA sequencing for subsets of tumors. Uveal melanoma is genomically distinct from other melanoma subtypes, harboring the lowest tumor mutation burden and with significantly mutated genes in the G-protein signaling pathway. Most cutaneous, acral, and mucosal melanomas share alterations in components of the MAPK, PI3K, p53, p16, and telomere pathways. However, the mechanism by which these pathways are activated or inactivated varies between melanoma subtypes. Additionally, we identify potential novel germline predisposition genes for some of the less common melanoma subtypes.
Significance: This is the largest whole-genome analysis of melanoma to date, comprehensively comparing the genomics of the four major melanoma subtypes. This study highlights both similarities and differences between the subtypes, providing insights into the etiology and biology of melanoma. This article is highlighted in the In This Issue feature, p. 2711.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
References
-
- Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017;545:175–80. - PubMed
-
- Wilmott JS, Johansson PA, Newell F, Waddell N, Ferguson P, Quek C, et al. Whole genome sequencing of melanomas in adolescent and young adults reveals distinct mutation landscapes and the potential role of germline variants in disease susceptibility. Int J Cancer 2019;144:1049–60. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
