

Genetic landscape of a large cohort of Primary Ovarian Insufficiency: New genes and pathways and implications for personalized medicine
- PMID: 36099812
- PMCID: PMC9475279
- DOI: 10.1016/j.ebiom.2022.104246
Genetic landscape of a large cohort of Primary Ovarian Insufficiency: New genes and pathways and implications for personalized medicine
Abstract
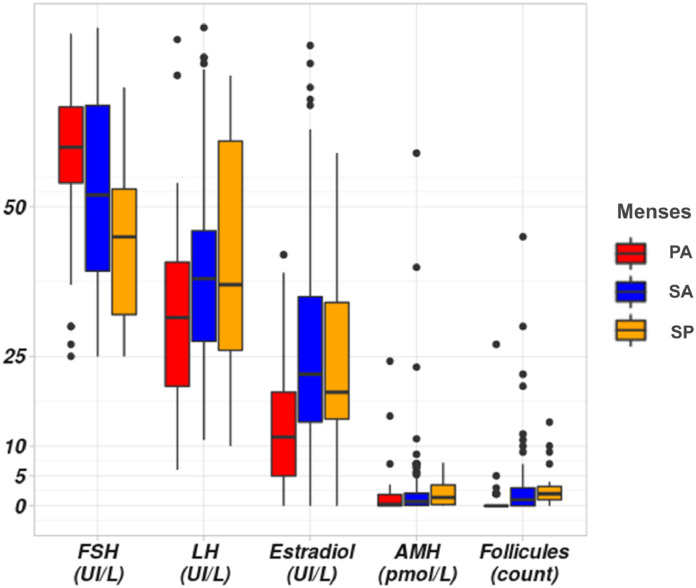
Background: Primary Ovarian Insufficiency (POI), a public health problem, affects 1-3.7% of women under 40 yielding infertility and a shorter lifespan. Most causes are unknown. Recently, genetic causes were identified, mostly in single families. We studied an unprecedented large cohort of POI to unravel its molecular pathophysiology.
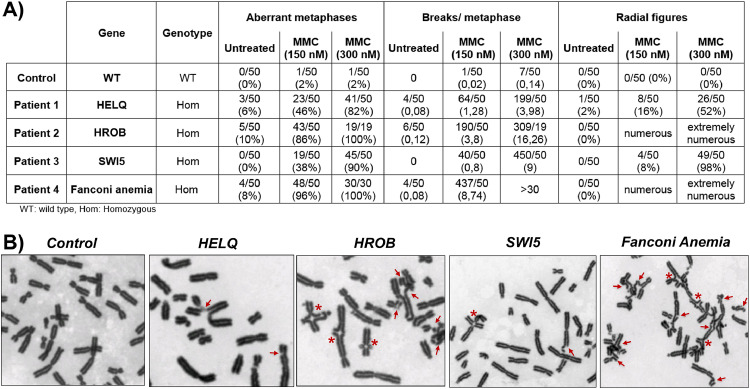
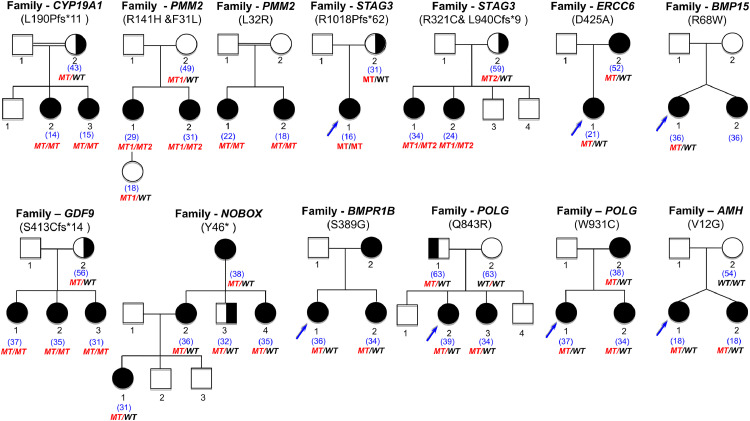
Methods: 375 patients with 70 families were studied using targeted (88 genes) or whole exome sequencing with pathogenic/likely-pathogenic variant selection. Mitomycin-induced chromosome breakages were studied in patients' lymphocytes if necessary.
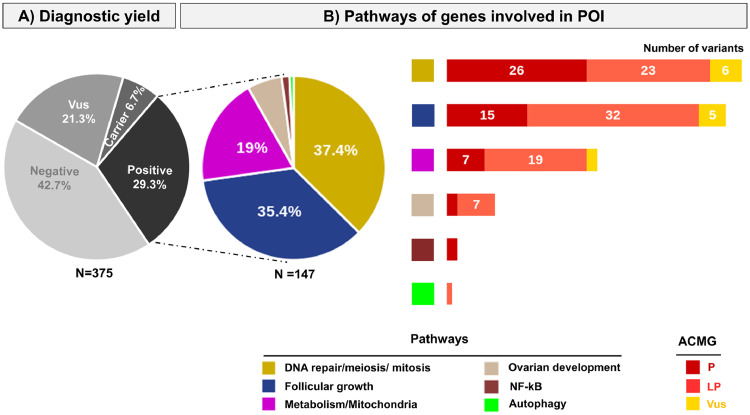
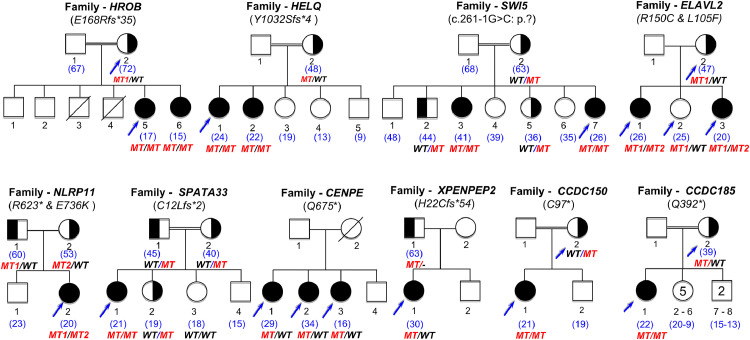
Findings: A high-yield of 29.3% supports a clinical genetic diagnosis of POI. In addition, we found strong evidence of pathogenicity for nine genes not previously related to a Mendelian phenotype or POI: ELAVL2, NLRP11, CENPE, SPATA33, CCDC150, CCDC185, including DNA repair genes: C17orf53(HROB), HELQ, SWI5 yielding high chromosomal fragility. We confirmed the causal role of BRCA2, FANCM, BNC1, ERCC6, MSH4, BMPR1A, BMPR1B, BMPR2, ESR2, CAV1, SPIDR, RCBTB1 and ATG7 previously reported in isolated patients/families. In 8.5% of cases, POI is the only symptom of a multi-organ genetic disease. New pathways were identified: NF-kB, post-translational regulation, and mitophagy (mitochondrial autophagy), providing future therapeutic targets. Three new genes have been shown to affect the age of natural menopause supporting a genetic link.
Interpretation: We have developed high-performance genetic diagnostic of POI, dissecting the molecular pathogenesis of POI and enabling personalized medicine to i) prevent/cure comorbidities for tumour/cancer susceptibility genes that could affect life-expectancy (37.4% of cases), or for genetically-revealed syndromic POI (8.5% of cases), ii) predict residual ovarian reserve (60.5% of cases). Genetic diagnosis could help to identify patients who may benefit from the promising in vitro activation-IVA technique in the near future, greatly improving its success in treating infertility.
Funding: Université Paris Saclay, Agence Nationale de Biomédecine.
Keywords: Meiosis/DNA repair genes; Mitophagy; NF-KB; Personalized medicine; Post - translational regulation; Primary ovarian insufficiency.
Copyright © 2022 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no conflict of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
