

Genetics and Pathogenesis of Parkinson's Syndrome
- PMID: 36100231
- PMCID: PMC10290758
- DOI: 10.1146/annurev-pathmechdis-031521-034145
Genetics and Pathogenesis of Parkinson's Syndrome
Abstract
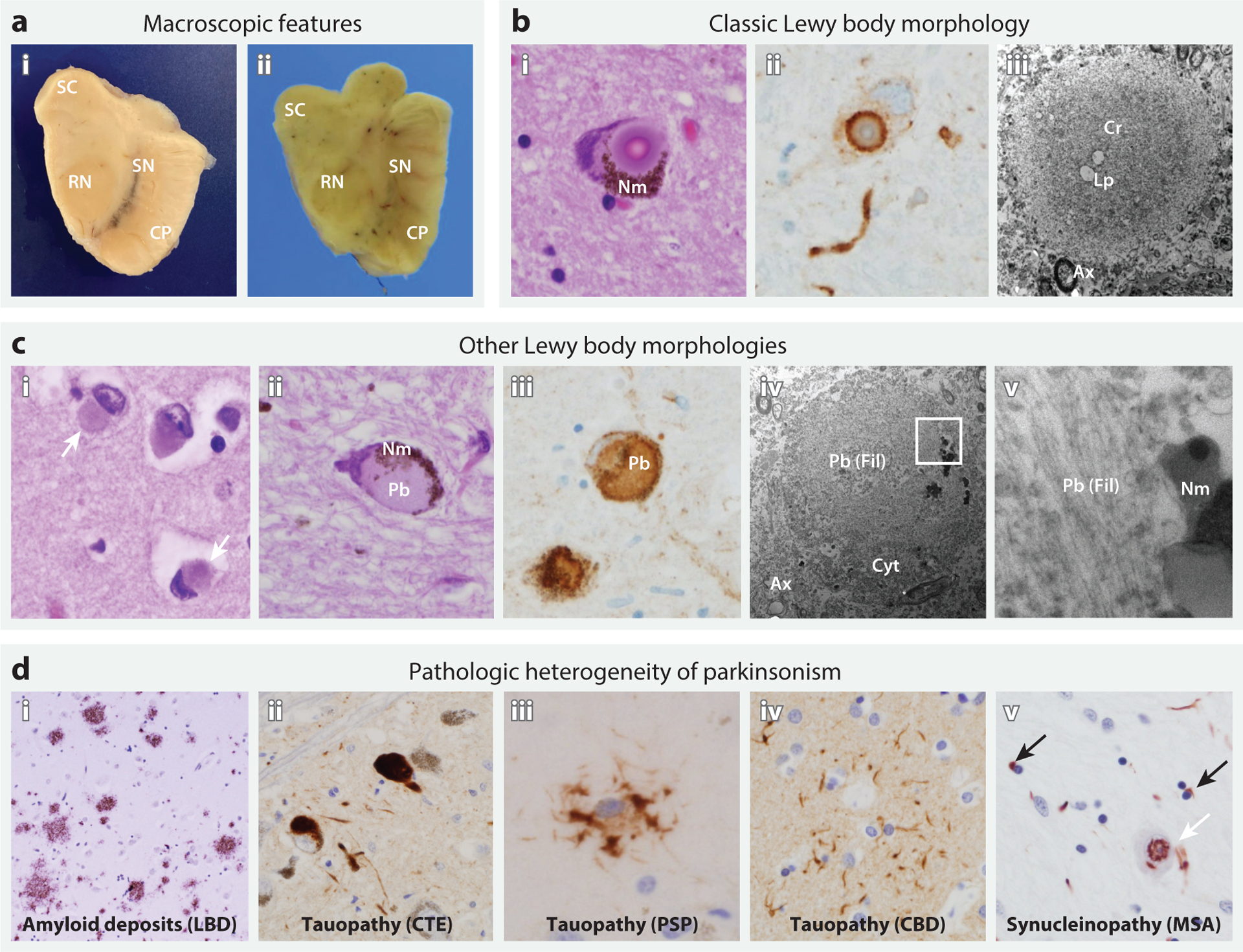
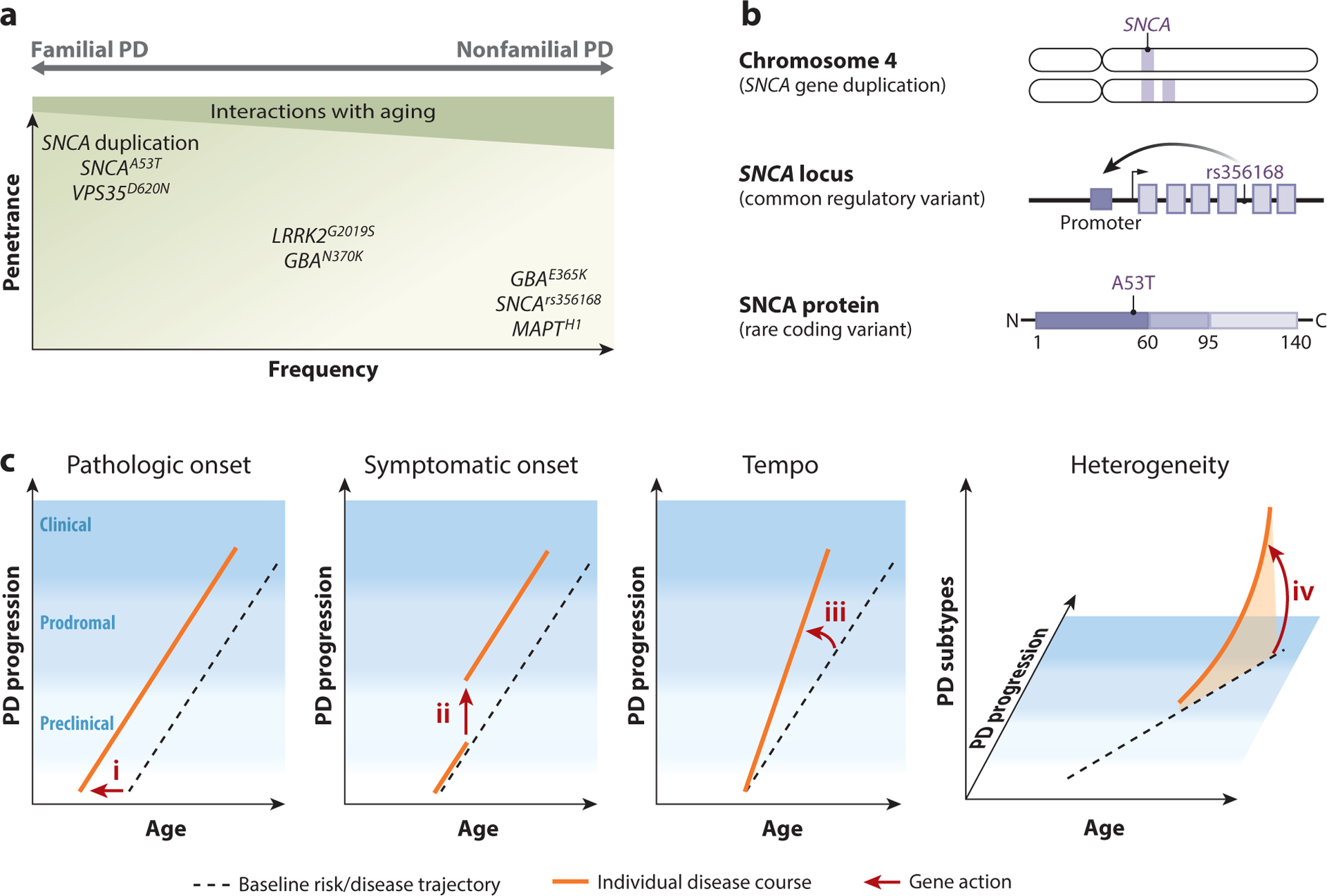
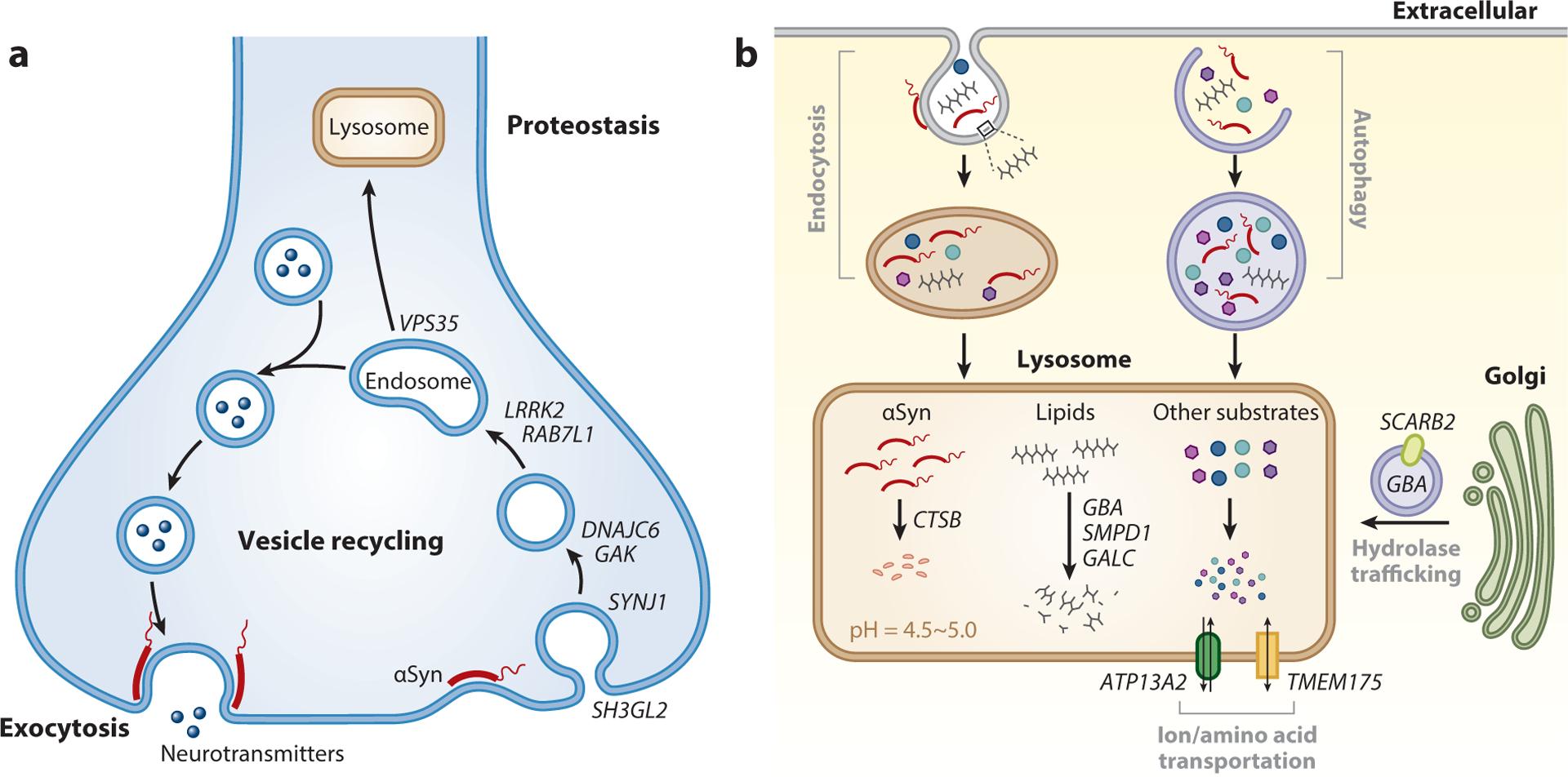
Parkinson's disease (PD) is clinically, pathologically, and genetically heterogeneous, resisting distillation to a single, cohesive disorder. Instead, each affected individual develops a virtually unique form of Parkinson's syndrome. Clinical manifestations consist of variable motor and nonmotor features, and myriad overlaps are recognized with other neurodegenerative conditions. Although most commonly characterized by alpha-synuclein protein pathology throughout the central and peripheral nervous systems, the distribution varies and other pathologies commonly modify PD or trigger similar manifestations. Nearly all PD is genetically influenced. More than 100 genes or genetic loci have been identified, and most cases likely arise from interactions among many common and rare genetic variants. Despite its complex architecture, insights from experimental genetic dissection coalesce to reveal unifying biological themes, including synaptic, lysosomal, mitochondrial, andimmune-mediated mechanisms of pathogenesis. This emerging understanding of Parkinson's syndrome, coupled with advances in biomarkers and targeted therapies, presages successful precision medicine strategies.
Keywords: GBA; LRRK2; Parkinson's disease; alpha-synuclein; functional genomics; heterogeneity; lysosome; mitochondria; oligogenic; parkinsonism; synapse.
Figures
References
-
- Weiner WJ. 2008. There is no Parkinson disease. Arch. Neurol 65(6):705–8 - PubMed
-
- Bloem BR, Okun MS, Klein C. 2021. Parkinson’s disease. Lancet 397(10291):2284–303 - PubMed
-
- Berg D, Borghammer P, Fereshtehnejad SM, Heinzel S, Horsager J, et al. 2021. Prodromal Parkinson disease subtypes—key to understanding heterogeneity. Nat. Rev. Neurol 17(6):349–61 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
