

Novel frameshift mutation in LIS1 gene is a probable cause of lissencephaly: a case report
- PMID: 36100855
- PMCID: PMC9472359
- DOI: 10.1186/s12887-022-03595-6
Novel frameshift mutation in LIS1 gene is a probable cause of lissencephaly: a case report
Abstract
Background: Lissencephaly (LIS) is a cortical malformation, characterized by smooth or nearly smooth cerebral surface and a shortage of gyral and sulcal development, which is caused by deficient neuronal migration during embryogenesis. Neuronal migration involves many gene products, among which is the product of the PAFAH1B1 gene, associated with this disease. LIS is a rare disease, characterized by low population frequency, and with non-specific clinical symptoms such as early epilepsy, developmental delay or cerebral palsy-like motor problems. Given that high-throughput sequencing techniques have been improving diagnosis, we have chosen this technique for addressing this patient.
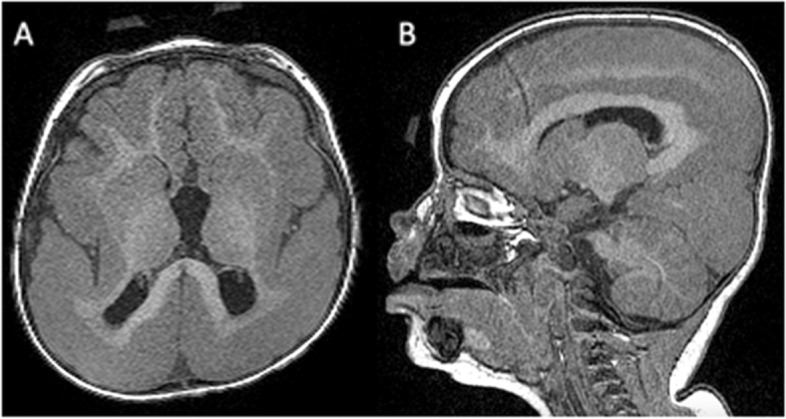
Case presentation: We present the case of a seven years old male patient with an undiagnosed rare disease, with non-specific clinical symptoms possibly compatible with lissencephaly. The patient was enrolled in a study that included the sequencing of his whole genome. Sequence data was analyzed following a bioinformatic pipeline. The variants obtained were annotated and then subjected to different filters for prioritization. Also mitochondrial genome was analyzed. A novel candidate frameshift insertion in known PAFAH1B1 gene was found, explaining the index case phenotype. The assessment through in silico tools reported that it causes nonsense mediated mechanisms and that it is damaging with high confidence scores. The insertion causes a change in the reading frame, and produces a premature stop codon, severely affecting the protein function and probably the silencing of one allele. The healthy mother did not carry the mutation, and the unaffected father was not available for analysis.
Conclusions: Through this work we found a novel de novo mutation in LIS1/PAFAH1B1 gene, as a likely cause of a rare disease in a young boy with non-specific clinical symptoms. The mutation found correlates with the phenotype studied since the loss of function in the gene product has already been described in this condition. Since there are no other variants in the PAFAH1B1 gene with low population frequency and due to family history, a de novo disease mechanism is proposed.
Keywords: Case report; Lissencephaly; Novel mutation; PAFAH1B1; Rare disease; Whole-genome sequencing.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
An isolated case of lissencephaly caused by the insertion of a mitochondrial genome-derived DNA sequence into the 5' untranslated region of the PAFAH1B1 (LIS1) gene.Hum Genomics. 2010 Aug;4(6):384-93. doi: 10.1186/1479-7364-4-6-384. Hum Genomics. 2010. PMID: 20846927 Free PMC article.
-
Lissencephaly in an epilepsy cohort: Molecular, radiological and clinical aspects.Eur J Paediatr Neurol. 2021 Jan;30:71-81. doi: 10.1016/j.ejpn.2020.12.011. Epub 2021 Jan 8. Eur J Paediatr Neurol. 2021. PMID: 33453472
-
Identification of a novel PAFAH1B1 missense mutation as a cause of mild lissencephaly with basal ganglia calcification.Brain Dev. 2019 Jan;41(1):29-35. doi: 10.1016/j.braindev.2018.07.009. Epub 2018 Aug 9. Brain Dev. 2019. PMID: 30100227
-
Genetic mechanisms underlying abnormal neuronal migration in classical lissencephaly.Trends Genet. 2007 Dec;23(12):623-30. doi: 10.1016/j.tig.2007.09.003. Epub 2007 Nov 8. Trends Genet. 2007. PMID: 17997185 Review.
-
[Molecular mechanism of lissencephaly--how LIS1 and NDEL1 regulate cytoplasmic dynein?].Brain Nerve. 2008 Apr;60(4):375-81. Brain Nerve. 2008. PMID: 18421979 Review. Japanese.
Cited by
-
Cryo-EM captures early intermediate steps in dynein activation by LIS1.Nat Commun. 2025 Aug 1;16(1):7054. doi: 10.1038/s41467-025-62185-z. Nat Commun. 2025. PMID: 40750582 Free PMC article.
-
Role of Physiotherapy in Pediatric Lissencephaly: A Case Report and Therapeutic Insights.Cureus. 2024 Jun 22;16(6):e62901. doi: 10.7759/cureus.62901. eCollection 2024 Jun. Cureus. 2024. PMID: 39040723 Free PMC article.
References
-
- Jones KL. Smith’s recognizable patterns of human malformation. 6. Philadelphia: Elsevier Saunders; 2006.
-
- Dobyns WB. Developmental aspects of lissencephaly and the lissencephaly syndromes. Birth Defects Orig Artic Ser. 1987;23(1):225–241. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
