

Transforming L1000 profiles to RNA-seq-like profiles with deep learning
- PMID: 36100892
- PMCID: PMC9472394
- DOI: 10.1186/s12859-022-04895-5
Transforming L1000 profiles to RNA-seq-like profiles with deep learning
Abstract
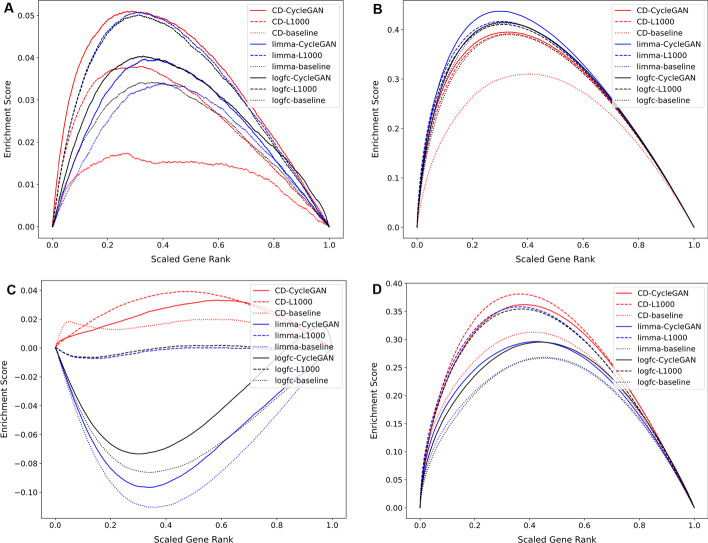
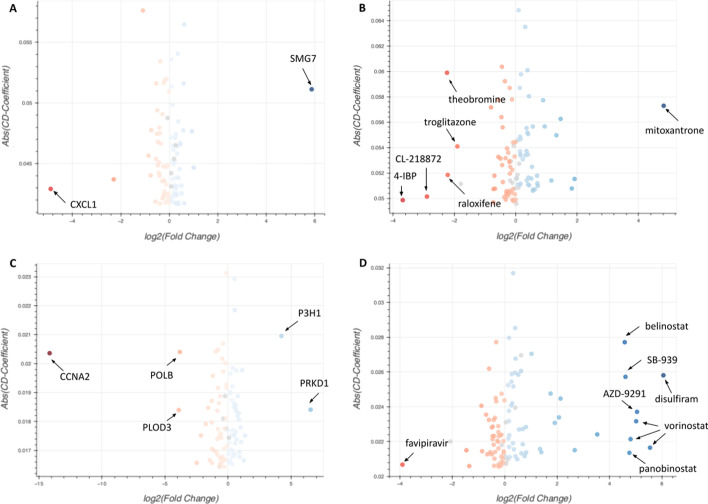
The L1000 technology, a cost-effective high-throughput transcriptomics technology, has been applied to profile a collection of human cell lines for their gene expression response to > 30,000 chemical and genetic perturbations. In total, there are currently over 3 million available L1000 profiles. Such a dataset is invaluable for the discovery of drug and target candidates and for inferring mechanisms of action for small molecules. The L1000 assay only measures the mRNA expression of 978 landmark genes while 11,350 additional genes are computationally reliably inferred. The lack of full genome coverage limits knowledge discovery for half of the human protein coding genes, and the potential for integration with other transcriptomics profiling data. Here we present a Deep Learning two-step model that transforms L1000 profiles to RNA-seq-like profiles. The input to the model are the measured 978 landmark genes while the output is a vector of 23,614 RNA-seq-like gene expression profiles. The model first transforms the landmark genes into RNA-seq-like 978 gene profiles using a modified CycleGAN model applied to unpaired data. The transformed 978 RNA-seq-like landmark genes are then extrapolated into the full genome space with a fully connected neural network model. The two-step model achieves 0.914 Pearson's correlation coefficients and 1.167 root mean square errors when tested on a published paired L1000/RNA-seq dataset produced by the LINCS and GTEx programs. The processed RNA-seq-like profiles are made available for download, signature search, and gene centric reverse search with unique case studies.
Keywords: Gene expression translation; Generative adversarial networks; L1000; RNA-seq.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures






References
-
- Zhu J-Y, Park T, Isola P, Efros AA. Unpaired image-to-image translation using cycle-consistent adversarial networks. In: 2017 IEEE International Conference on Computer Vision (ICCV) 2017.
-
- Goodfellow IJ, Pouget-Abadie J, Mirza M, Xu B, Warde-Farley D, Ozair S, Courville A, Bengio Y. Generative adversarial networks. arXiv [statML] 2014.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
