

Characterization of phenylalanine hydroxylase gene variants and analysis of genotype-phenotype correlation in patients with phenylalanine hydroxylase deficiency from Fujian Province, Southeastern China
- PMID: 36104584
- PMCID: PMC9618490
- DOI: 10.1007/s11033-022-07579-8
Characterization of phenylalanine hydroxylase gene variants and analysis of genotype-phenotype correlation in patients with phenylalanine hydroxylase deficiency from Fujian Province, Southeastern China
Abstract
Background: Phenylalanine hydroxylase deficiency (PAHD) is the most prevalent inherited disorder of amino acid metabolism in China. Its complex phenotype includes many variants and genotypes among different populations.
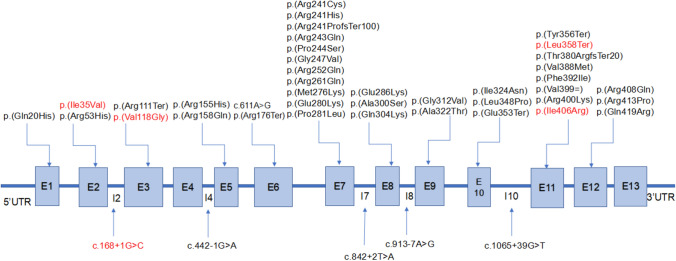
Methods and results: In this study, we analyzed the phenylalanine hydroxylase gene (PAH) variants in a cohort of 93 PAHD patients from Fujian Province. We also assessed genotype and phenotype correlation in patients with PAHD. A total of 44 different pathogenic variants were identified, including five novel variants. The three most prevalent variants among all patents were c.158G > A, p.(Arg53His) (18.03%), c.721C > T, p.(Arg241Cys) (14.75%), and c.728G > A, p.(Arg243Gln) (7.65%). The frequency of the c.158G > A, p.(Arg53His) variant was highest in patients with mild hyperphenylalaninemia, whereas the frequency of the c.1197A > T, p.(Val399 =) and c.331C > T, p.(Arg111Ter) variants was highest in patients with classic phenylketonuria. The most abundant genotypes observed in PAHD patients were c.[158G > A];[728G > A], c.[158G > A];[442-1G > A], and c.[158G > A];[721C > T]. Comparing allelic phenotype to genotypic phenotype values yielded fairly accurate predictions of phenotype, with an overall consistency rate was 85.71% for PAHD patients.
Conclusions: Our study identified a PAH variant spectrum in PAHD patients from Fujian Province, Southeastern China. Quantitative correlation analysis between genotype and phenotype severity is helpful for genetic counseling and management.
Keywords: Genotype–phenotype correlation; Phenylalanine hydroxylase deficiency; Prediction; Southeastern China; Variant spectrum.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
