

Phenotype Analysis of Fused in Sarcoma Mutations in Amyotrophic Lateral Sclerosis
- PMID: 36105853
- PMCID: PMC9469212
- DOI: 10.1212/NXG.0000000000200011
Phenotype Analysis of Fused in Sarcoma Mutations in Amyotrophic Lateral Sclerosis
Abstract
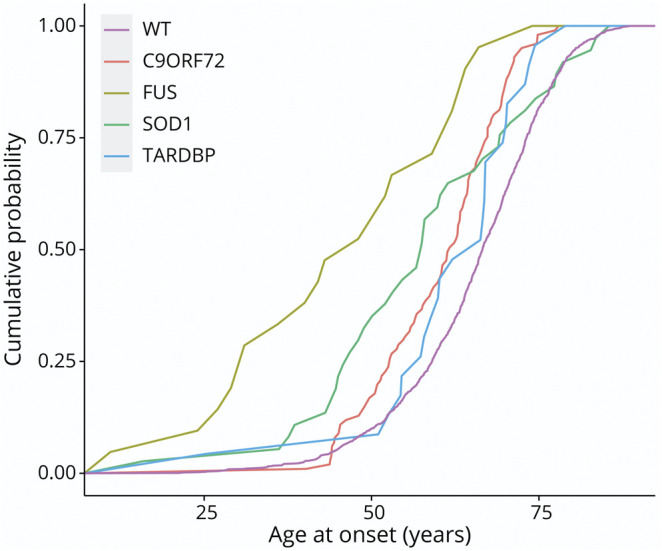
Background and objectives: Pathogenic variations in fused in sarcoma (FUS) are among the most common genetic causes of amyotrophic lateral sclerosis (ALS) worldwide. They are supposedly characterized by a homogeneous pure motor phenotype with early-onset and short disease duration. However, a few FUS-mutated cases with a very late disease onset and slow progression have been reported. To analyze genotype-phenotype correlations and identify the prognostic factors in FUS-ALS cases.
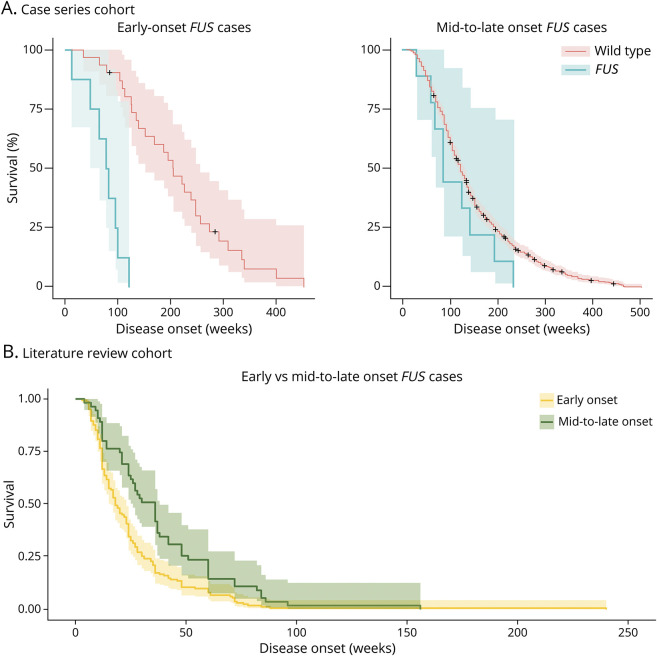
Methods: We identified and cross-sectionally analyzed 22 FUS-ALS patient histories from a single-center cohort of 2,615 genetically tested patients and reviewed 289 previously published FUS-ALS cases. Survival analysis was performed by Kaplan-Meier survival curves, followed by the log-rank test and multivariate Cox analysis.
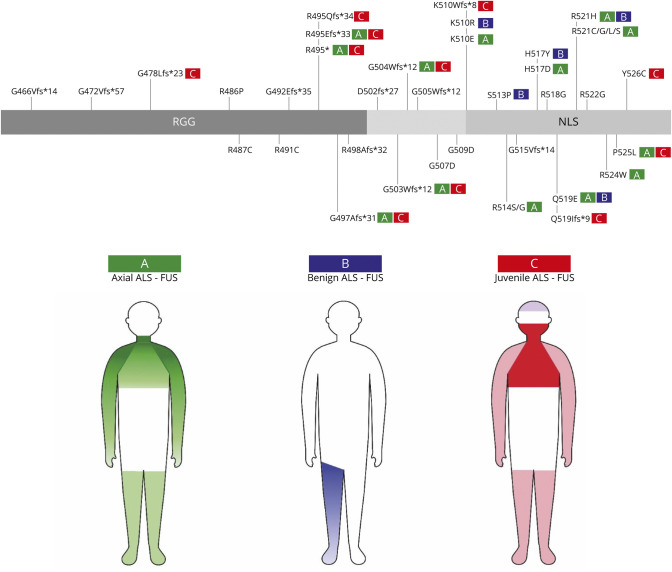
Results: Survival of FUS-ALS is age-dependent: In our cohort, early-onset cases had a rapid disease progression and short survival (p = 0.000003) while the outcome of FUS-mutated patients with mid-to-late onset did not differ from non-FUS-ALS patients (p = 0.437). Meta-analysis of literature data confirmed this trend (p = 0.00003). This survival pattern is not observed in other ALS-related genes in our series. We clustered FUS-ALS patients in 3 phenotypes: (1) axial ALS, with upper cervical and dropped-head onset in mid-to-late adulthood; (2) benign ALS, usually with a late-onset and slow disease progression; and (3) juvenile ALS, often with bulbar onset and preceded by learning disability or mild mental retardation. Those phenotypes arise from different mutations.
Discussion: We observed specific genotype-phenotype correlations of FUS-ALS and identified age at onset as the most critical prognostic factor. Our results demonstrated that FUS mutations underlie a specific subtype of ALS and enable a careful stratification of newly diagnosed FUS-ALS cases for clinical course and potential therapeutic windows. This will be crucial in the light of incoming gene-specific therapy.
Copyright © 2022 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures
References
-
- Kwiatkowski TJ, Bosco DA, LeClerc AL, et al. . Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205-1208. - PubMed
-
- Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2017;88(7):540-549. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous