

Molecular epidemiology and antibiotic resistance profiles of invasive Haemophilus influenzae from Norway 2017-2021
- PMID: 36106084
- PMCID: PMC9467436
- DOI: 10.3389/fmicb.2022.973257
Molecular epidemiology and antibiotic resistance profiles of invasive Haemophilus influenzae from Norway 2017-2021
Abstract
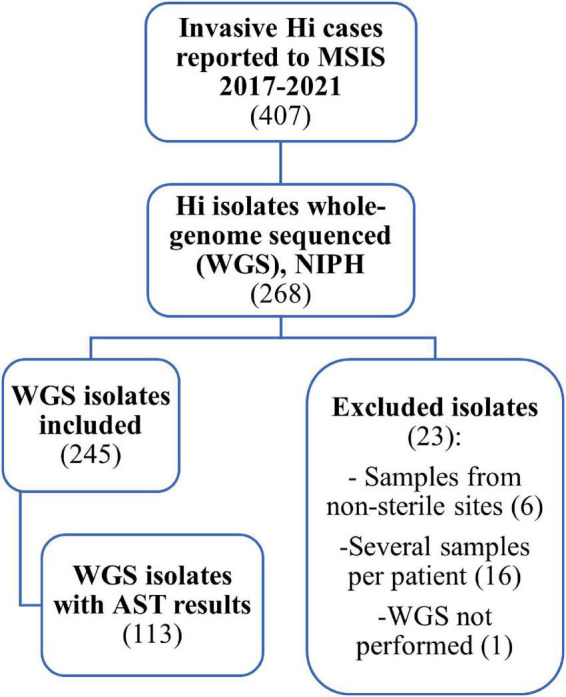
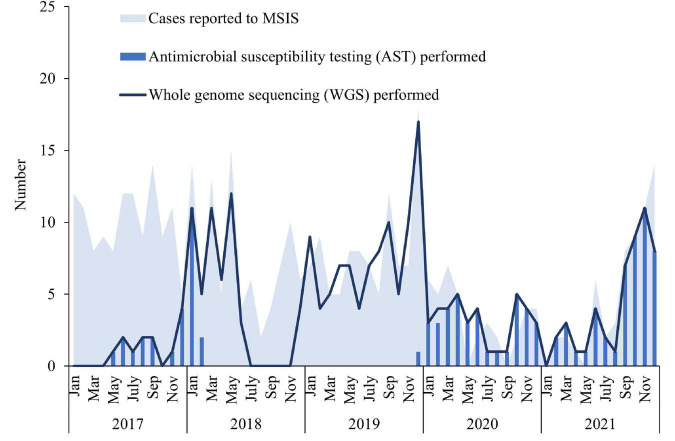
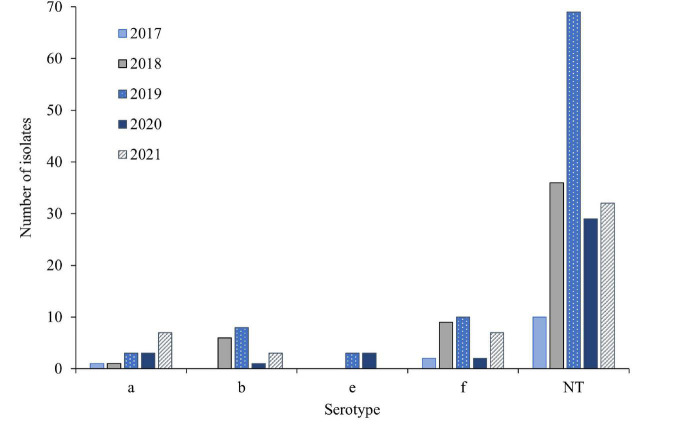
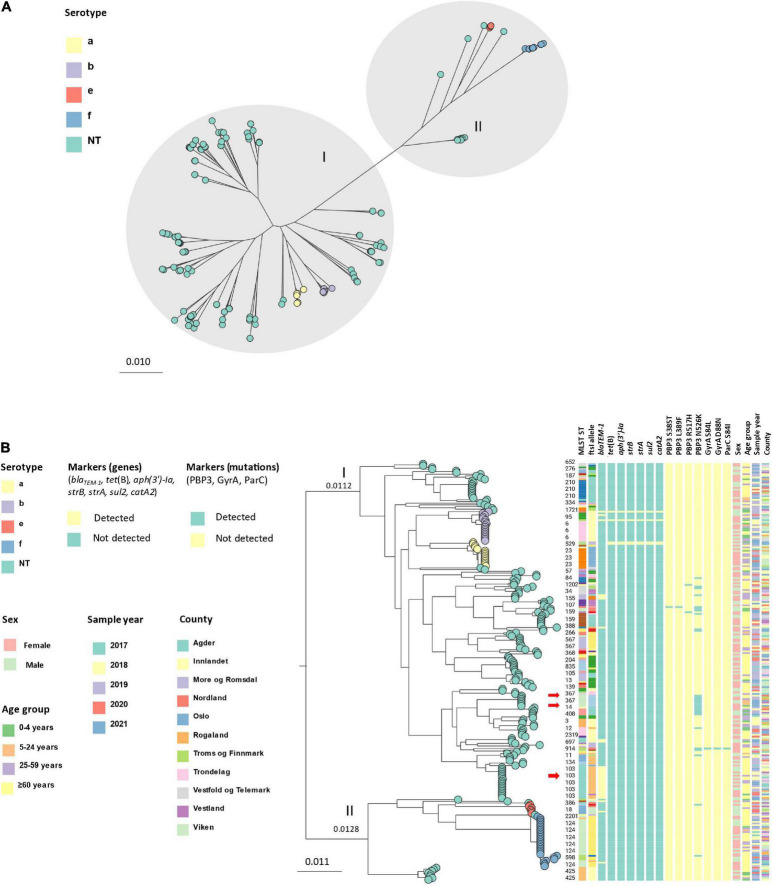
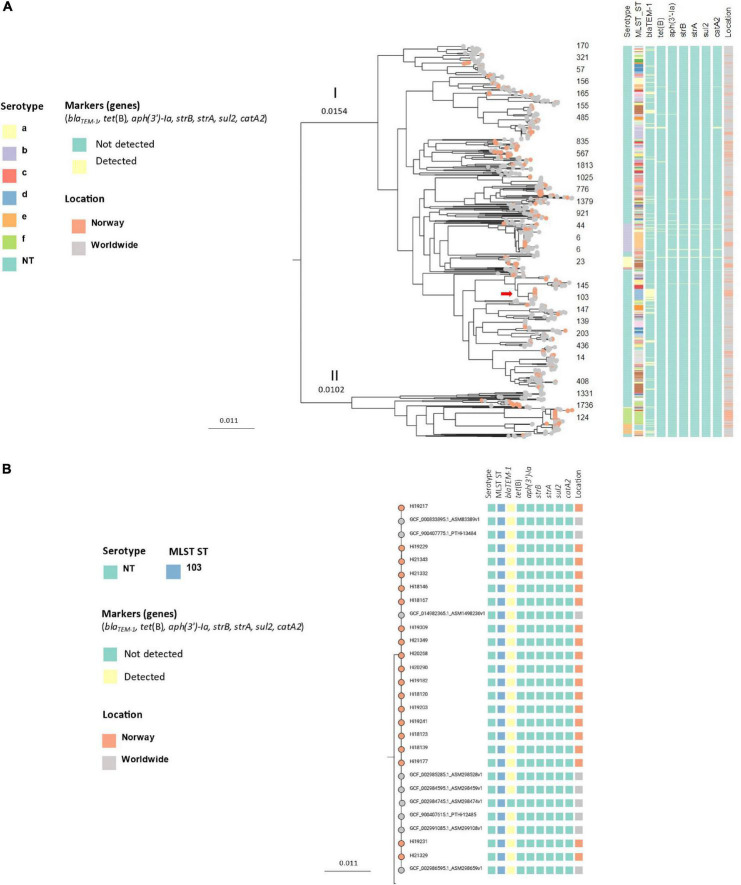
Invasive Haemophilus influenzae (Hi) disease has decreased in countries that included Hi type b (Hib) vaccination in their childhood immunization programs in the 1990s. Non-typeable (NT) and non-b strains are now the leading causes of invasive Hi disease in Europe, with most cases reported in young children and the elderly. Concerningly, no vaccines toward such strains are available and beta-lactam resistance is increasing. We describe the epidemiology of invasive Hi disease reported to the Norwegian Surveillance System for Communicable Diseases (MSIS) (2017-2021, n = 407). Whole-genome sequencing (WGS) was performed on 245 isolates. We investigated the molecular epidemiology (core genome phylogeny) and the presence of antibiotic resistance markers (including chromosomal mutations associated with beta-lactam or quinolone resistance). For isolates characterized with both WGS and phenotypic antibiotic susceptibility testing (AST) (n = 113) we assessed correlation between resistance markers and susceptibility categorization by calculation of sensitivity, specificity, and predictive values. Incidence rates of invasive Hi disease in Norway ranged from 0.7 to 2.3 per 100,000 inhabitants/year (mean 1.5 per 100,000) and declined during the COVID-19 pandemic. The bacterial population consisted of two major phylogenetic groups with subclustering by serotype and multi-locus sequence type (ST). NTHi accounted for 71.8% (176). The distribution of STs was in line with previous European reports. We identified 13 clusters, including four encapsulated and three previously described international NTHi clones with bla TEM-1 (ST103) or altered PBP3 (rPBP3) (ST14/IIA and ST367/IIA). Resistance markers were detected in 25.3% (62/245) of the isolates, with bla TEM-1 (31, 50.0%) and rPBP3 (28, 45.2%) being the most frequent. All isolates categorized as resistant to aminopenicillins, tetracycline or chloramphenicol possessed relevant resistance markers, and the absence of relevant substitutions in PBP3 and GyrA/ParC predicted susceptibility to cefotaxime, ceftriaxone, meropenem and quinolones. Among the 132 WGS-only isolates, one isolate had PBP3 substitutions associated with resistance to third-generation cephalosporins, and one isolate had GyrA/ParC alterations associated with quinolone resistance. The detection of international virulent and resistant NTHi clones underlines the need for a global molecular surveillance system. WGS is a useful supplement to AST and should be performed on all invasive isolates.
Keywords: Haemophilus influenzae (Hi); Norway; antibiotic resistance; epidemiology; invasive; surveillance; vaccine; whole-genome sequencing (WGS).
Copyright © 2022 Tønnessen, García, Debech, Lindstrøm, Wester and Skaare.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous