

Von Hippel-Lindau disease: insights into oxygen sensing, protein degradation, and cancer
- PMID: 36106637
- PMCID: PMC9479583
- DOI: 10.1172/JCI162480
Von Hippel-Lindau disease: insights into oxygen sensing, protein degradation, and cancer
Abstract
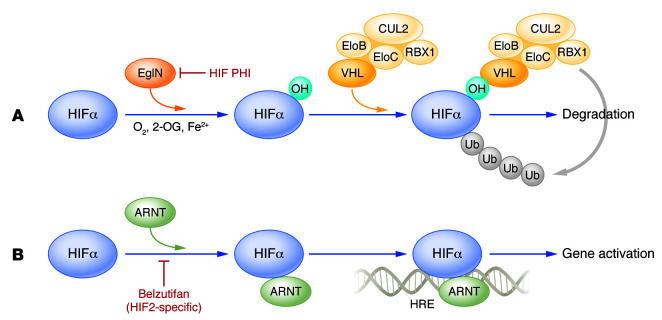
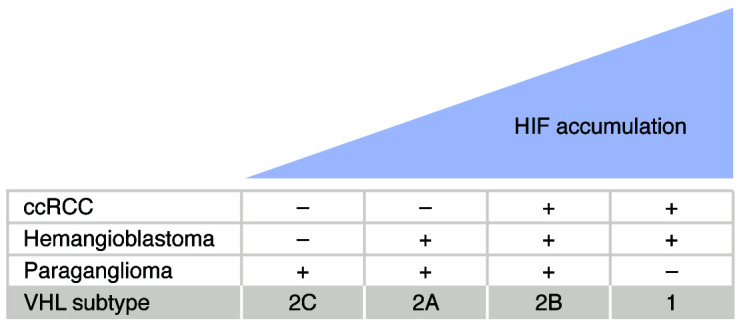
Germline loss-of-function mutations of the VHL tumor suppressor gene cause von Hippel-Lindau disease, which is associated with an increased risk of hemangioblastomas, clear cell renal cell carcinomas (ccRCCs), and paragangliomas. This Review describes mechanisms involving the VHL gene product in oxygen sensing, protein degradation, and tumor development and current therapeutic strategies targeting these mechanisms. The VHL gene product is the substrate recognition subunit of a ubiquitin ligase that targets the α subunit of the heterodimeric hypoxia-inducible factor (HIF) transcription factor for proteasomal degradation when oxygen is present. This oxygen dependence stems from the requirement that HIFα be prolyl-hydroxylated on one (or both) of two conserved prolyl residues by members of the EglN (also called PHD) prolyl hydroxylase family. Deregulation of HIF, and particularly HIF2, drives the growth of VHL-defective ccRCCs. Drugs that inhibit the HIF-responsive gene product VEGF are now mainstays of ccRCC treatment. An allosteric HIF2 inhibitor was recently approved for the treatment of ccRCCs arising in the setting of VHL disease and has advanced to phase III testing for sporadic ccRCCs based on promising phase I/II data. Orally available EglN inhibitors are being tested for the treatment of anemia and ischemia. Five of these agents have been approved for the treatment of anemia in the setting of chronic kidney disease in various countries around the world.
Conflict of interest statement
Figures
References
-
- Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22(24):409–5004. - PubMed
