

Glutamine prevents acute kidney injury by modulating oxidative stress and apoptosis in tubular epithelial cells
- PMID: 36107633
- PMCID: PMC9675453
- DOI: 10.1172/jci.insight.163161
Glutamine prevents acute kidney injury by modulating oxidative stress and apoptosis in tubular epithelial cells
Abstract
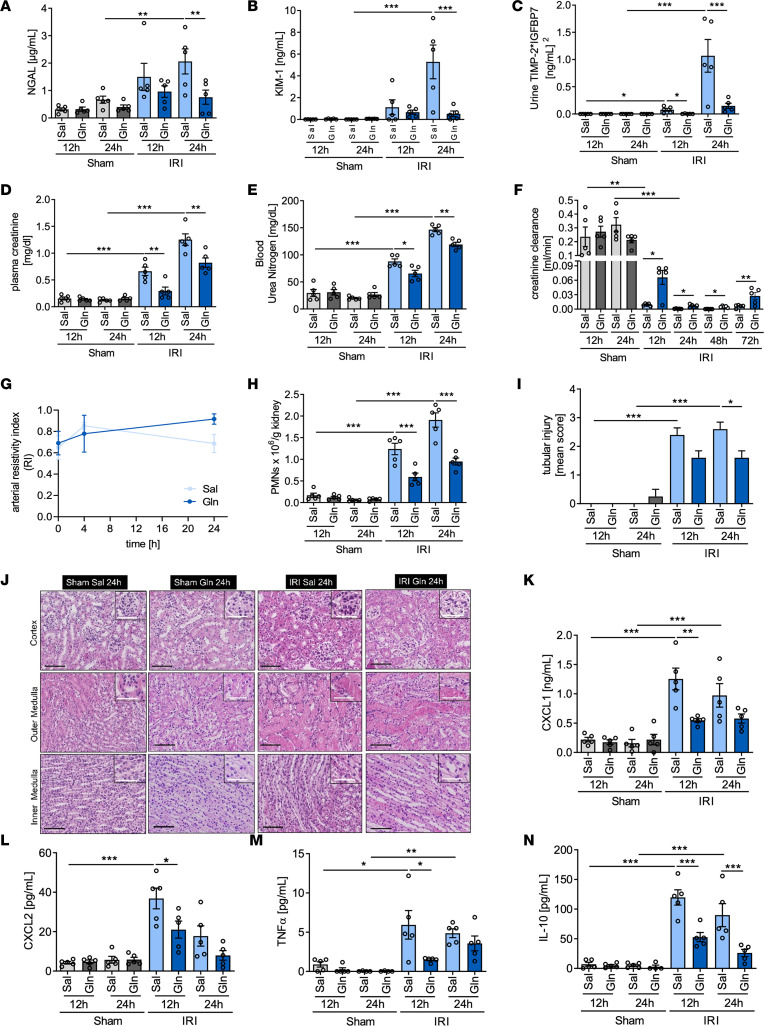
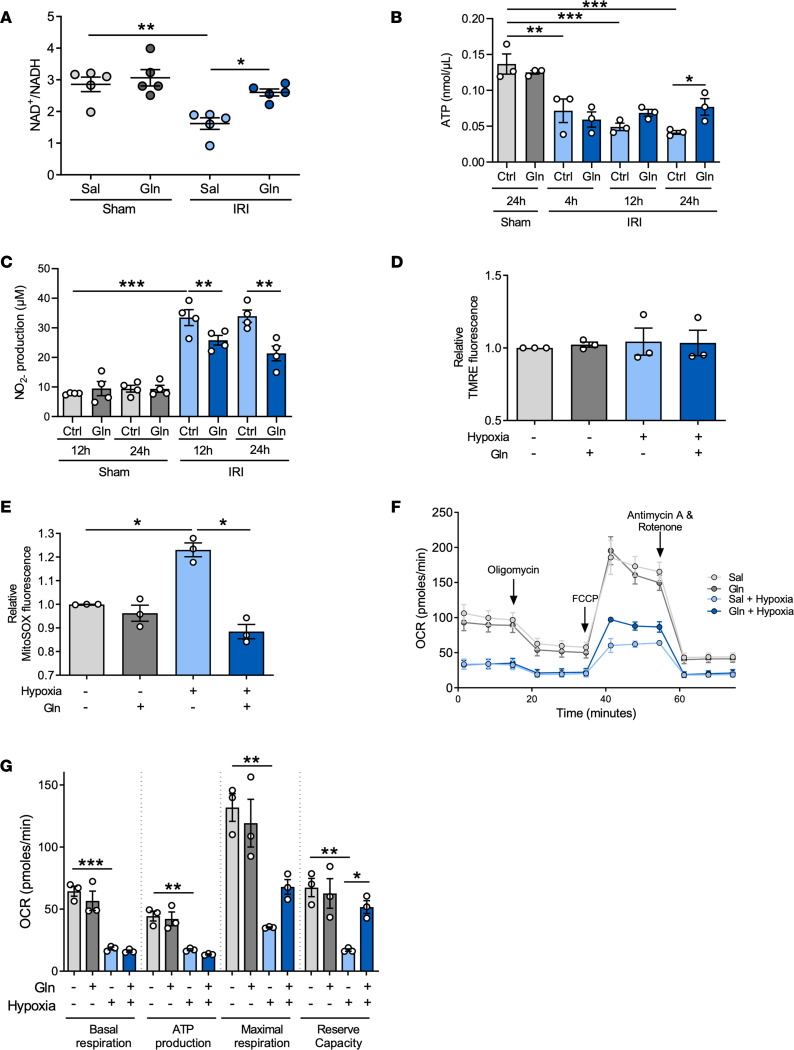
Acute kidney injury (AKI) represents a common complication in critically ill patients that is associated with increased morbidity and mortality. In a murine AKI model induced by ischemia/reperfusion injury (IRI), we show that glutamine significantly decreases kidney damage and improves kidney function. We demonstrate that glutamine causes transcriptomic and proteomic reprogramming in murine renal tubular epithelial cells (TECs), resulting in decreased epithelial apoptosis, decreased neutrophil recruitment, and improved mitochondrial functionality and respiration provoked by an ameliorated oxidative phosphorylation. We identify the proteins glutamine gamma glutamyltransferase 2 (Tgm2) and apoptosis signal-regulating kinase (Ask1) as the major targets of glutamine in apoptotic signaling. Furthermore, the direct modulation of the Tgm2-HSP70 signalosome and reduced Ask1 activation resulted in decreased JNK activation, leading to diminished mitochondrial intrinsic apoptosis in TECs. Glutamine administration attenuated kidney damage in vivo during AKI and TEC viability in vitro under inflammatory or hypoxic conditions.
Keywords: Apoptosis; Cellular immune response; Immunology; Mitochondria; Nephrology.
Conflict of interest statement
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
