

Trajectory Surface Hopping for a Polarizable Embedding QM/MM Formulation
- PMID: 36107729
- PMCID: PMC9527758
- DOI: 10.1021/acs.jpca.2c04756
Trajectory Surface Hopping for a Polarizable Embedding QM/MM Formulation
Abstract
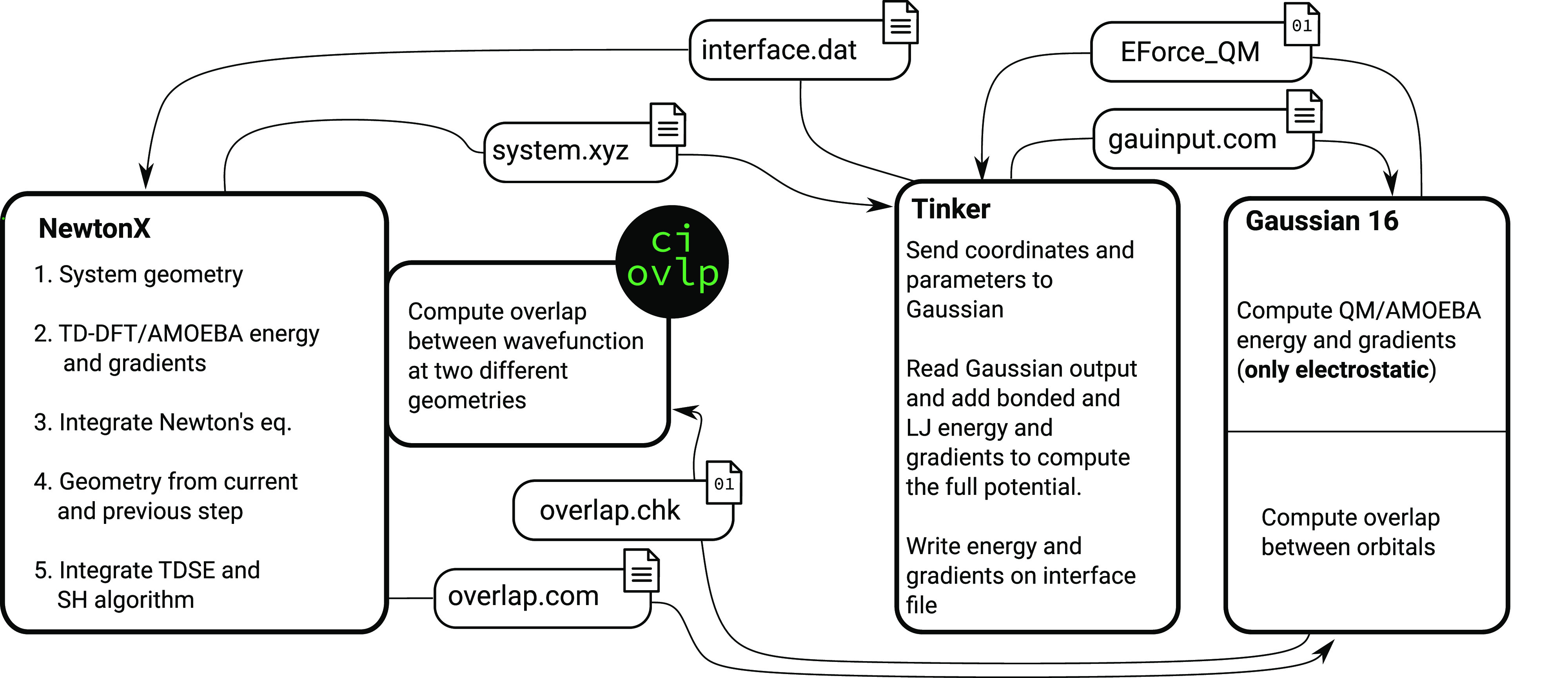
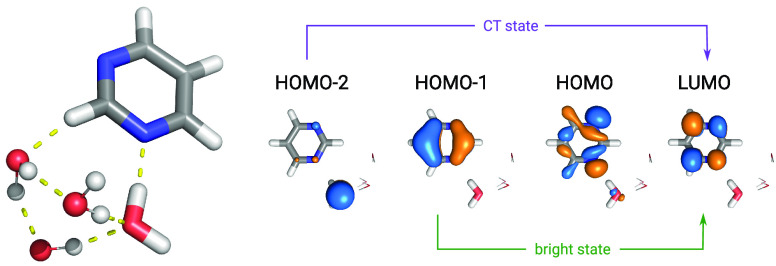
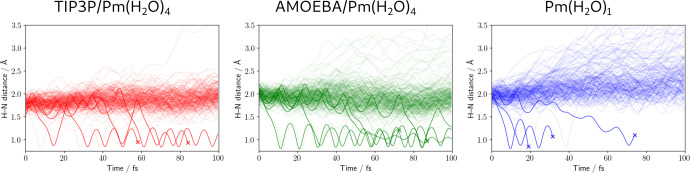
We present the implementation of trajectory surface-hopping nonadiabatic dynamics for a polarizable embedding QM/MM formulation. Time-dependent density functional theory was used at the quantum mechanical level of theory, whereas the molecular mechanics description involved the polarizable AMOEBA force field. This implementation has been obtained by integrating the surface-hopping program Newton-X NS with an interface between the Gaussian 16 and the Tinker suites of codes to calculate QM/AMOEBA energies and forces. The implementation has been tested on a photoinduced electron-driven proton-transfer reaction involving pyrimidine and a hydrogen-bonded water surrounded by a small cluster of water molecules and within a large water droplet.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
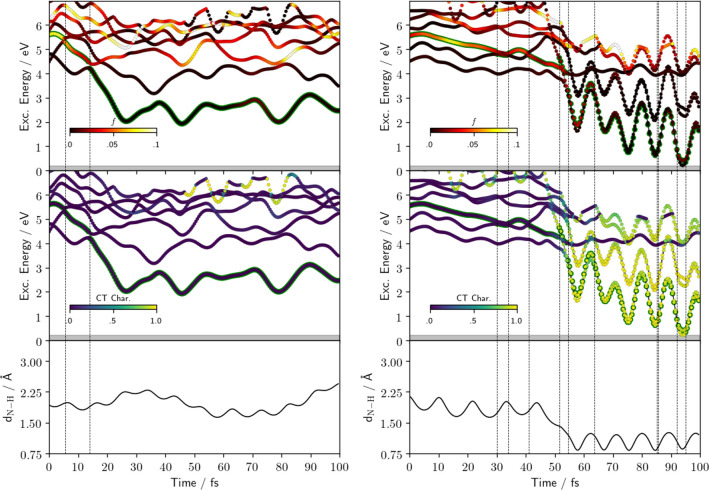
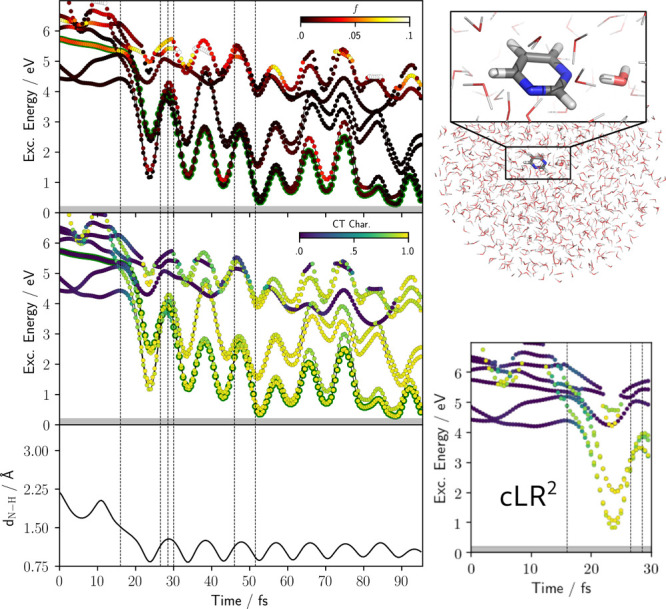
 character of the states. (Bottom) Distance
of Pm N atom from the hydrogen atom of the initially H-bonded water
molecule.
character of the states. (Bottom) Distance
of Pm N atom from the hydrogen atom of the initially H-bonded water
molecule.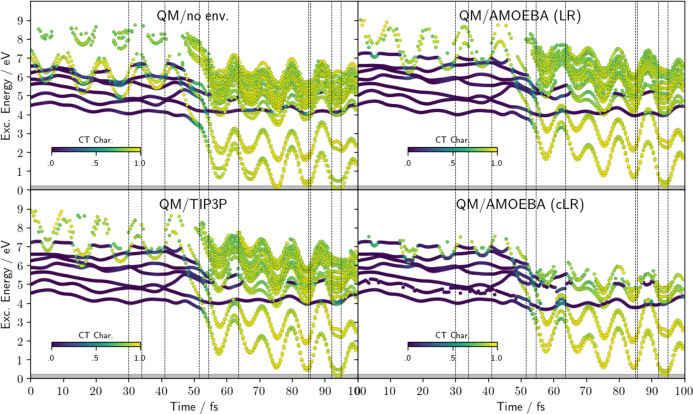
References
-
- Persico M.; Granucci G. An overview of nonadiabatic dynamics simulations methods, with focus on the direct approach versus the fitting of potential energy surfaces. Theor. Chem. Acc. 2014, 133, 1526.10.1007/s00214-014-1526-1. - DOI
-
- Tully J. C. Molecular dynamics with electronic transitions. J. Chem. Phys. 1990, 93, 1061–1071. 10.1063/1.459170. - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
