

Robust deep learning-based protein sequence design using ProteinMPNN
- PMID: 36108050
- PMCID: PMC9997061
- DOI: 10.1126/science.add2187
Robust deep learning-based protein sequence design using ProteinMPNN
Abstract
Although deep learning has revolutionized protein structure prediction, almost all experimentally characterized de novo protein designs have been generated using physically based approaches such as Rosetta. Here, we describe a deep learning-based protein sequence design method, ProteinMPNN, that has outstanding performance in both in silico and experimental tests. On native protein backbones, ProteinMPNN has a sequence recovery of 52.4% compared with 32.9% for Rosetta. The amino acid sequence at different positions can be coupled between single or multiple chains, enabling application to a wide range of current protein design challenges. We demonstrate the broad utility and high accuracy of ProteinMPNN using x-ray crystallography, cryo-electron microscopy, and functional studies by rescuing previously failed designs, which were made using Rosetta or AlphaFold, of protein monomers, cyclic homo-oligomers, tetrahedral nanoparticles, and target-binding proteins.
Conflict of interest statement
Figures
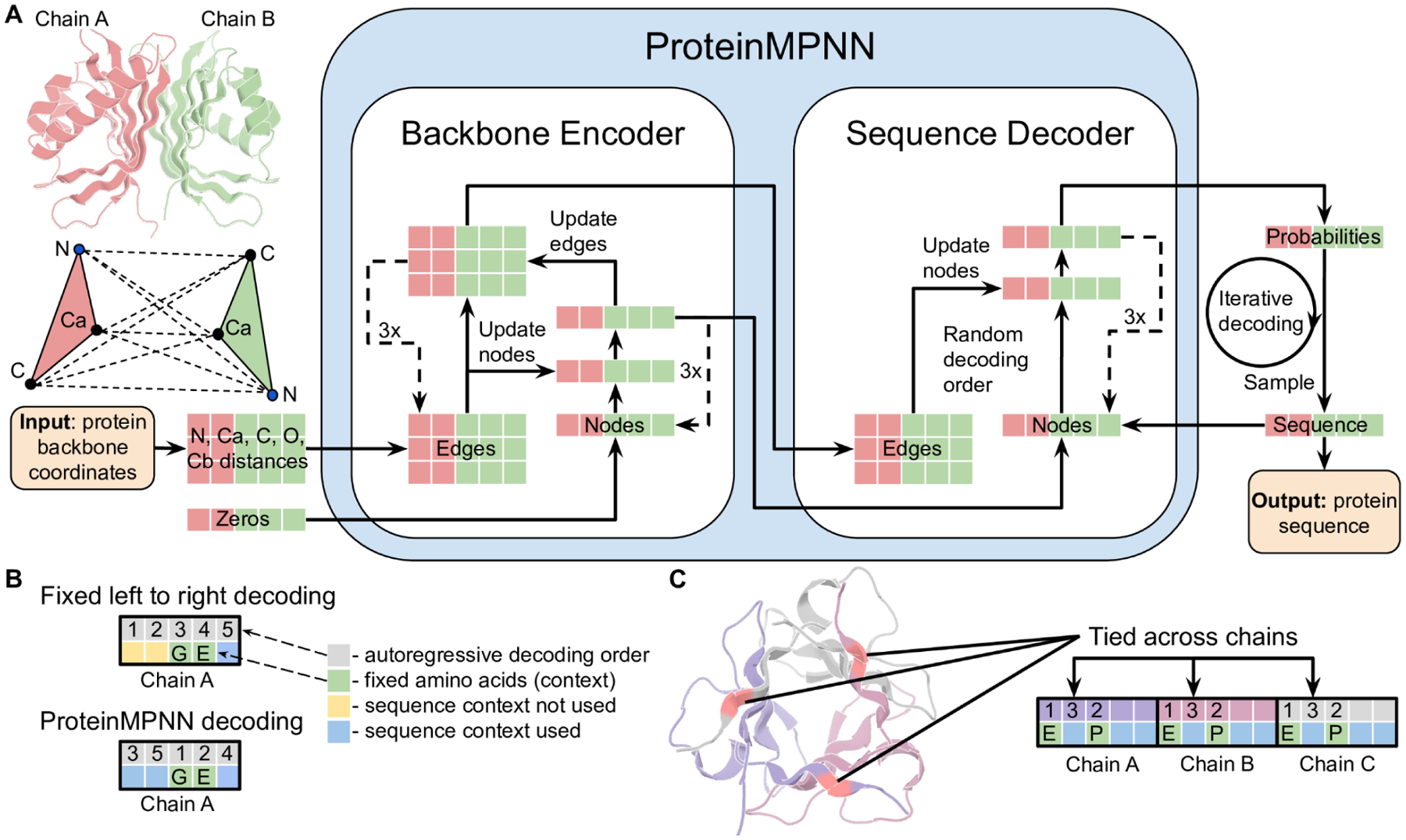
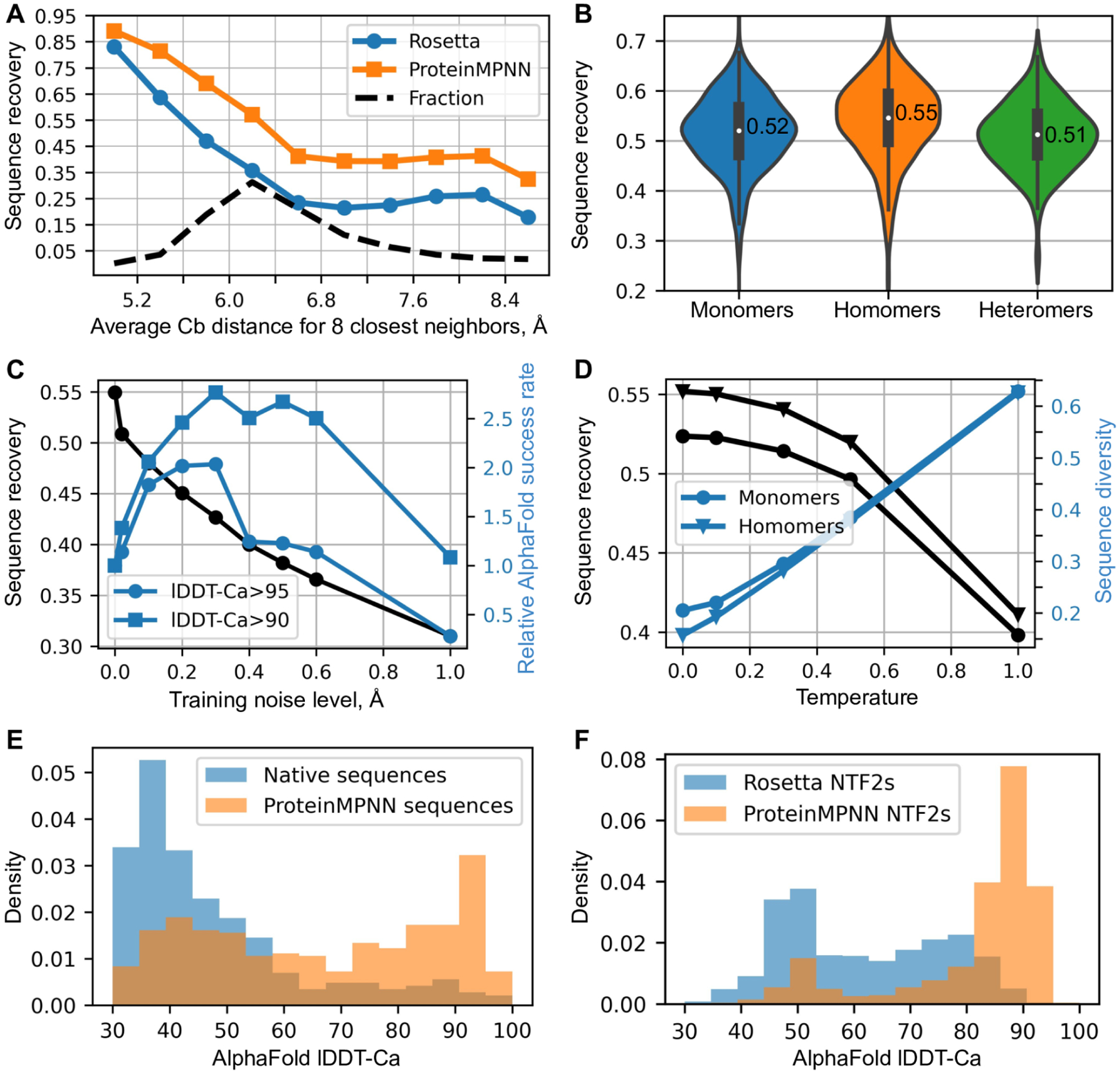
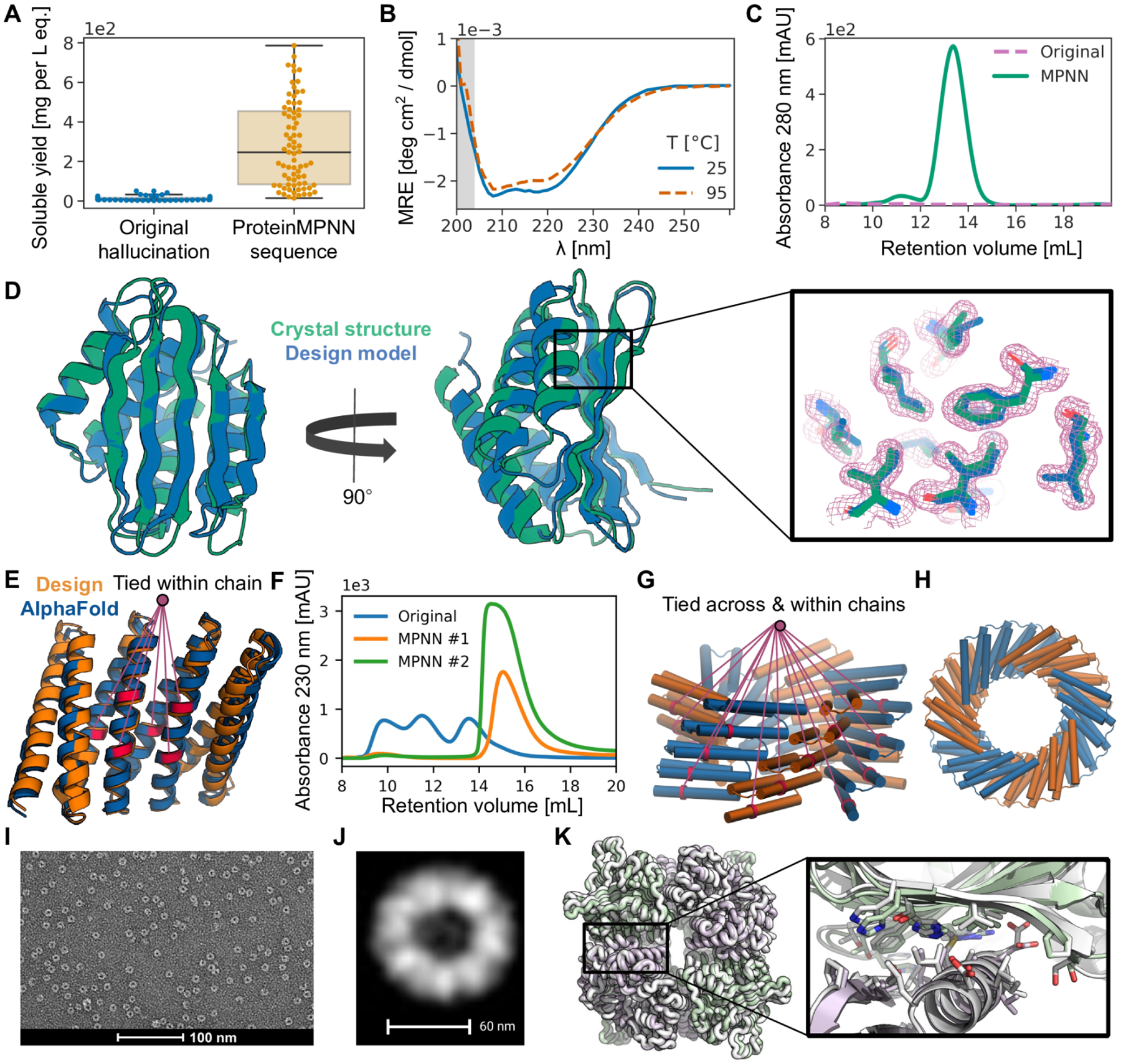
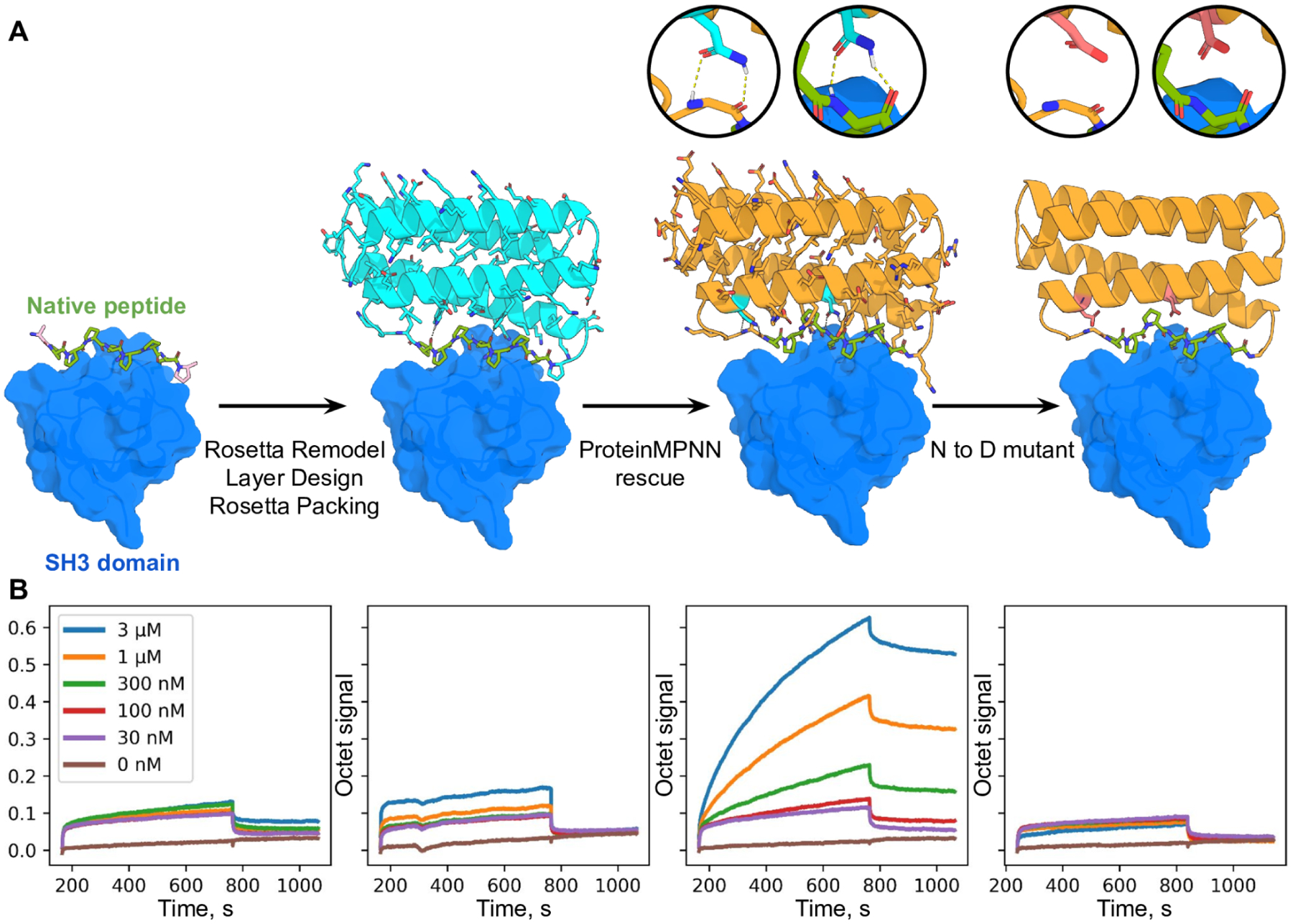
References
-
- Ingraham J, Garg V, Barzilay R, & Jaakkola T (2019). Generative models for graph-based protein design. Advances in Neural Information Processing Systems, 32.
-
- Qi Y, & Zhang JZ (2020). DenseCPD: improving the accuracy of neural-network-based computational protein sequence design with DenseNet. Journal of Chemical Information and Modeling, 60(3), 1245–1252. - PubMed
-
- Jing B, Eismann S, Suriana P, Townshend RJL, & Dror R (2020, September). Learning from Protein Structure with Geometric Vector Perceptrons. In International Conference on Learning Representations.
-
- Strokach A, Becerra D, Corbi-Verge C, Perez-Riba A, & Kim PM (2020). Fast and flexible protein design using deep graph neural networks. Cell systems, 11(4), 402–411. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
