

Inflammatory signalling in atrial cardiomyocytes: a novel unifying principle in atrial fibrillation pathophysiology
- PMID: 36109633
- PMCID: PMC9477170
- DOI: 10.1038/s41569-022-00759-w
Inflammatory signalling in atrial cardiomyocytes: a novel unifying principle in atrial fibrillation pathophysiology
Abstract
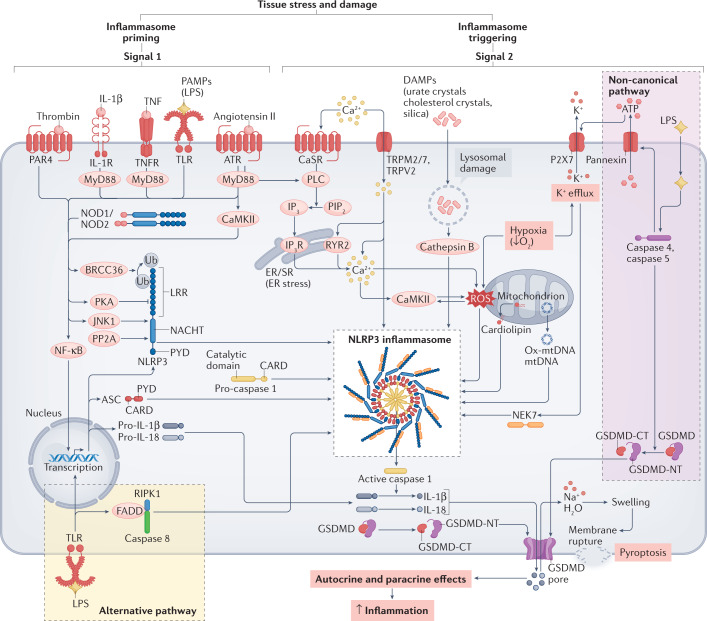
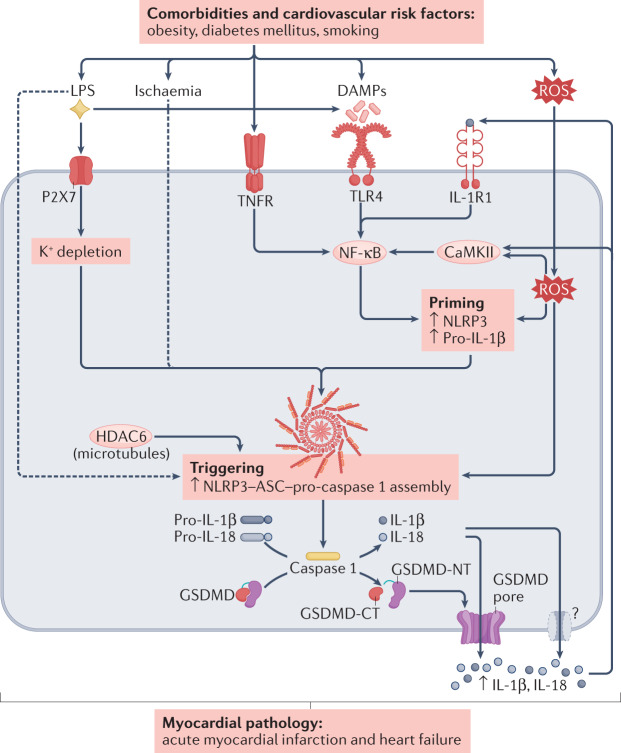
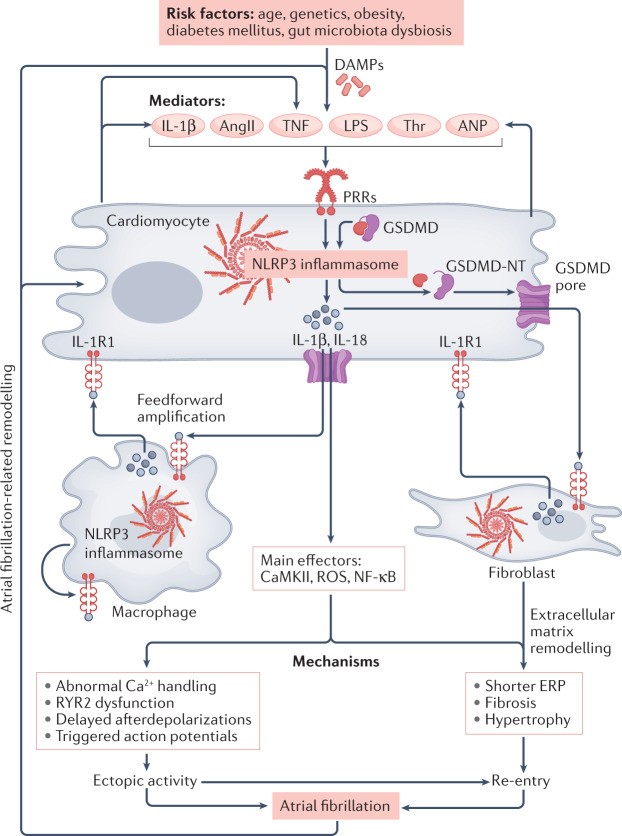
Inflammation has been implicated in atrial fibrillation (AF), a very common and clinically significant cardiac rhythm disturbance, but its precise role remains poorly understood. Work performed over the past 5 years suggests that atrial cardiomyocytes have inflammatory signalling machinery - in particular, components of the NLRP3 (NACHT-, LRR- and pyrin domain-containing 3) inflammasome - that is activated in animal models and patients with AF. Furthermore, work in animal models suggests that NLRP3 inflammasome activation in atrial cardiomyocytes might be a sufficient and necessary condition for AF occurrence. In this Review, we evaluate the evidence for the role and pathophysiological significance of cardiomyocyte NLRP3 signalling in AF. We first summarize the evidence for a role of inflammation in AF and review the biochemical properties of the NLRP3 inflammasome, as defined primarily in studies of classic inflammation. We then briefly consider the broader evidence for a role of inflammatory signalling in heart disease, particularly conditions that predispose individuals to develop AF. We provide a detailed discussion of the available information about atrial cardiomyocyte NLRP3 inflammasome signalling in AF and related conditions and evaluate the possibility that similar signalling might be important in non-myocyte cardiac cells. We then review the evidence on the role of active resolution of inflammation and its potential importance in suppressing AF-related inflammatory signalling. Finally, we consider the therapeutic potential and broader implications of this new knowledge and highlight crucial questions to be addressed in future research.
© 2022. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
