

Inferring structural and dynamical properties of gene networks from data with deep learning
- PMID: 36110897
- PMCID: PMC9469930
- DOI: 10.1093/nargab/lqac068
Inferring structural and dynamical properties of gene networks from data with deep learning
Abstract
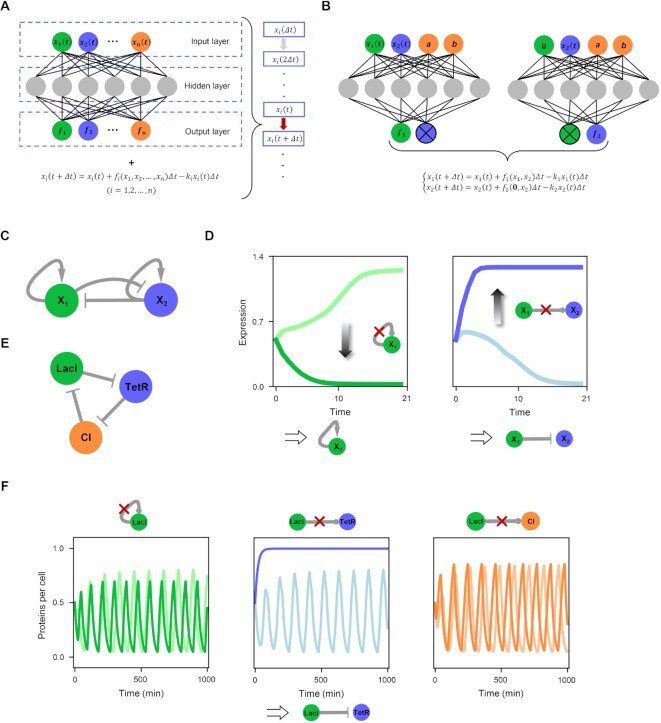
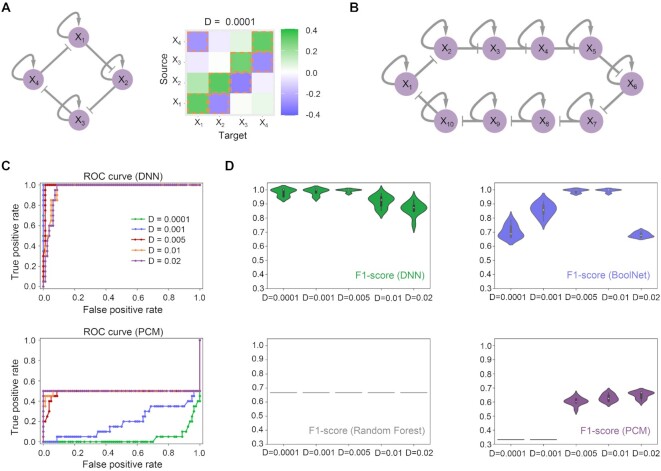
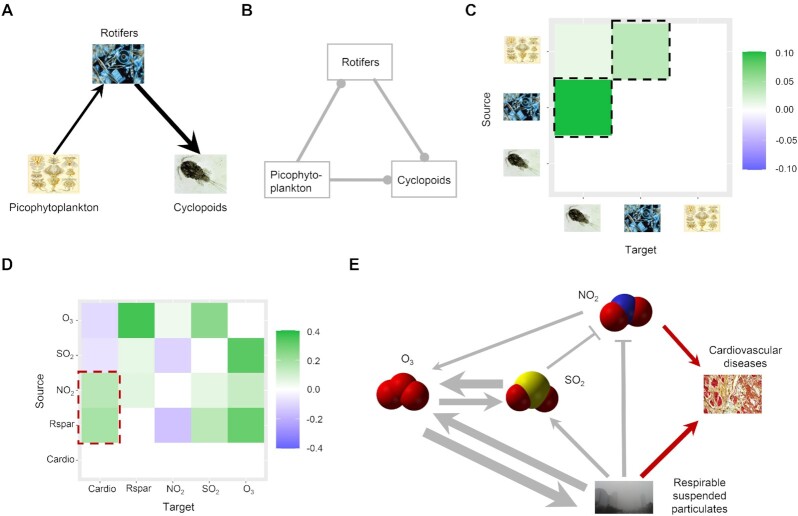
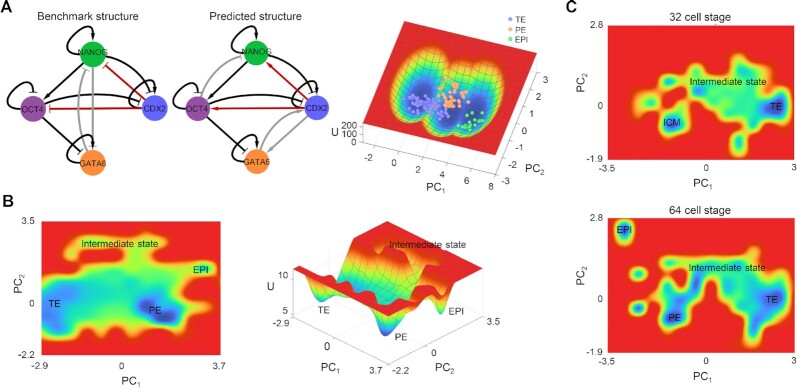
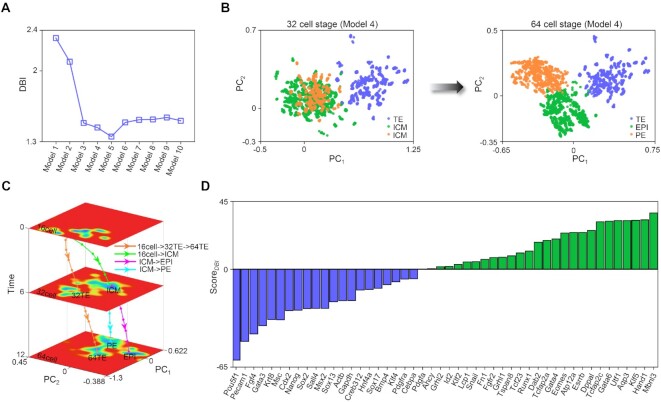
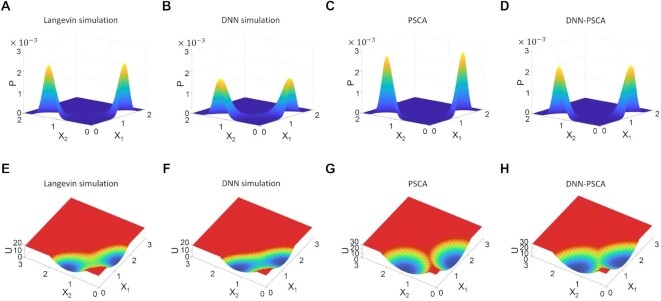
The reconstruction of gene regulatory networks (GRNs) from data is vital in systems biology. Although different approaches have been proposed to infer causality from data, some challenges remain, such as how to accurately infer the direction and type of interactions, how to deal with complex network involving multiple feedbacks, as well as how to infer causality between variables from real-world data, especially single cell data. Here, we tackle these problems by deep neural networks (DNNs). The underlying regulatory network for different systems (gene regulations, ecology, diseases, development) can be successfully reconstructed from trained DNN models. We show that DNN is superior to existing approaches including Boolean network, Random Forest and partial cross mapping for network inference. Further, by interrogating the ensemble DNN model trained from single cell data from dynamical system perspective, we are able to unravel complex cell fate dynamics during preimplantation development. We also propose a data-driven approach to quantify the energy landscape for gene regulatory systems, by combining DNN with the partial self-consistent mean field approximation (PSCA) approach. We anticipate the proposed method can be applied to other fields to decipher the underlying dynamical mechanisms of systems from data.
© The Author(s) 2022. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures
References
LinkOut - more resources
Full Text Sources
