

Oxygen toxicity: cellular mechanisms in normobaric hyperoxia
- PMID: 36112262
- PMCID: PMC9483325
- DOI: 10.1007/s10565-022-09773-7
Oxygen toxicity: cellular mechanisms in normobaric hyperoxia
Abstract
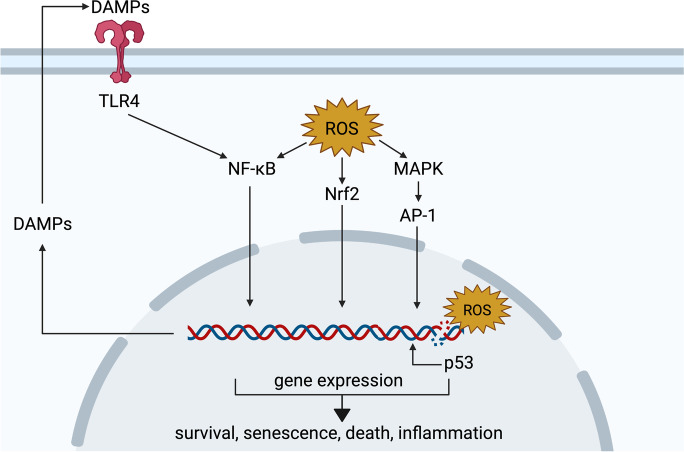
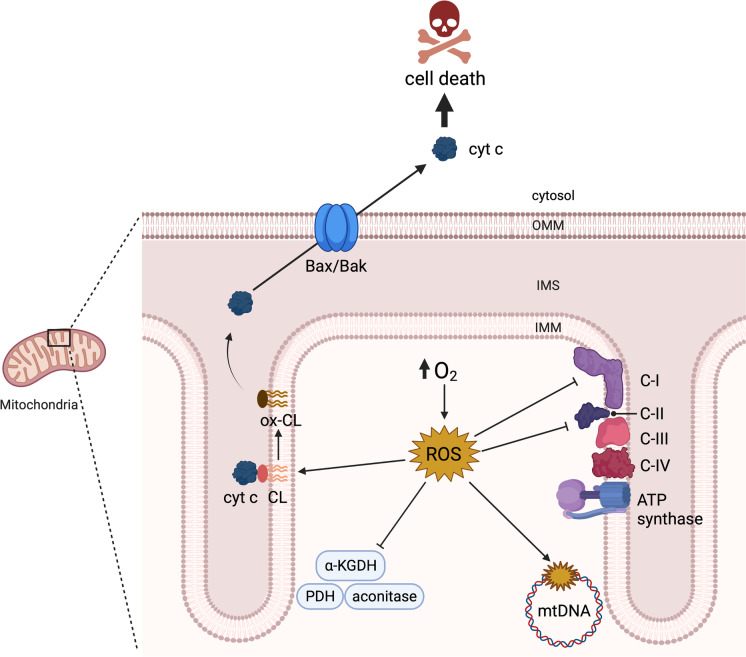
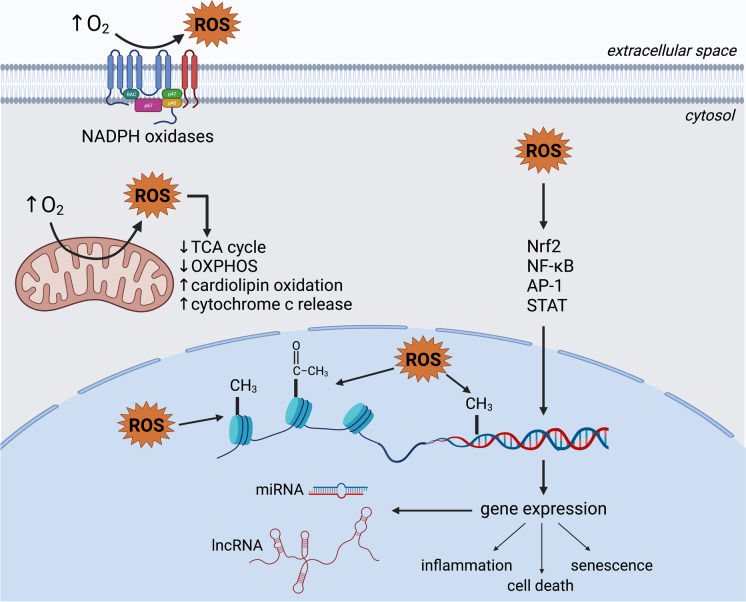
In clinical settings, oxygen therapy is administered to preterm neonates and to adults with acute and chronic conditions such as COVID-19, pulmonary fibrosis, sepsis, cardiac arrest, carbon monoxide poisoning, and acute heart failure. In non-clinical settings, divers and astronauts may also receive supplemental oxygen. In addition, under current standard cell culture practices, cells are maintained in atmospheric oxygen, which is several times higher than what most cells experience in vivo. In all the above scenarios, the elevated oxygen levels (hyperoxia) can lead to increased production of reactive oxygen species from mitochondria, NADPH oxidases, and other sources. This can cause cell dysfunction or death. Acute hyperoxia injury impairs various cellular functions, manifesting ultimately as physiological deficits. Chronic hyperoxia, particularly in the neonate, can disrupt development, leading to permanent deficiencies. In this review, we discuss the cellular activities and pathways affected by hyperoxia, as well as strategies that have been developed to ameliorate injury. • Hyperoxia promotes overproduction of reactive oxygen species (ROS). • Hyperoxia dysregulates a variety of signaling pathways, such as the Nrf2, NF-κB and MAPK pathways. • Hyperoxia causes cell death by multiple pathways. • Antioxidants, particularly, mitochondria-targeted antioxidants, have shown promising results as therapeutic agents against oxygen toxicity in animal models.
Keywords: Antioxidants; Cell death; Hyperoxia; Mitochondria; Oxygen toxicity; Reactive oxygen species.
© 2022. The Author(s), under exclusive licence to Springer Nature B.V.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
