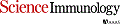DPP9 deficiency: An inflammasomopathy that can be rescued by lowering NLRP1/IL-1 signaling
- PMID: 36112693
- PMCID: PMC9844213
- DOI: 10.1126/sciimmunol.abi4611
DPP9 deficiency: An inflammasomopathy that can be rescued by lowering NLRP1/IL-1 signaling
Abstract
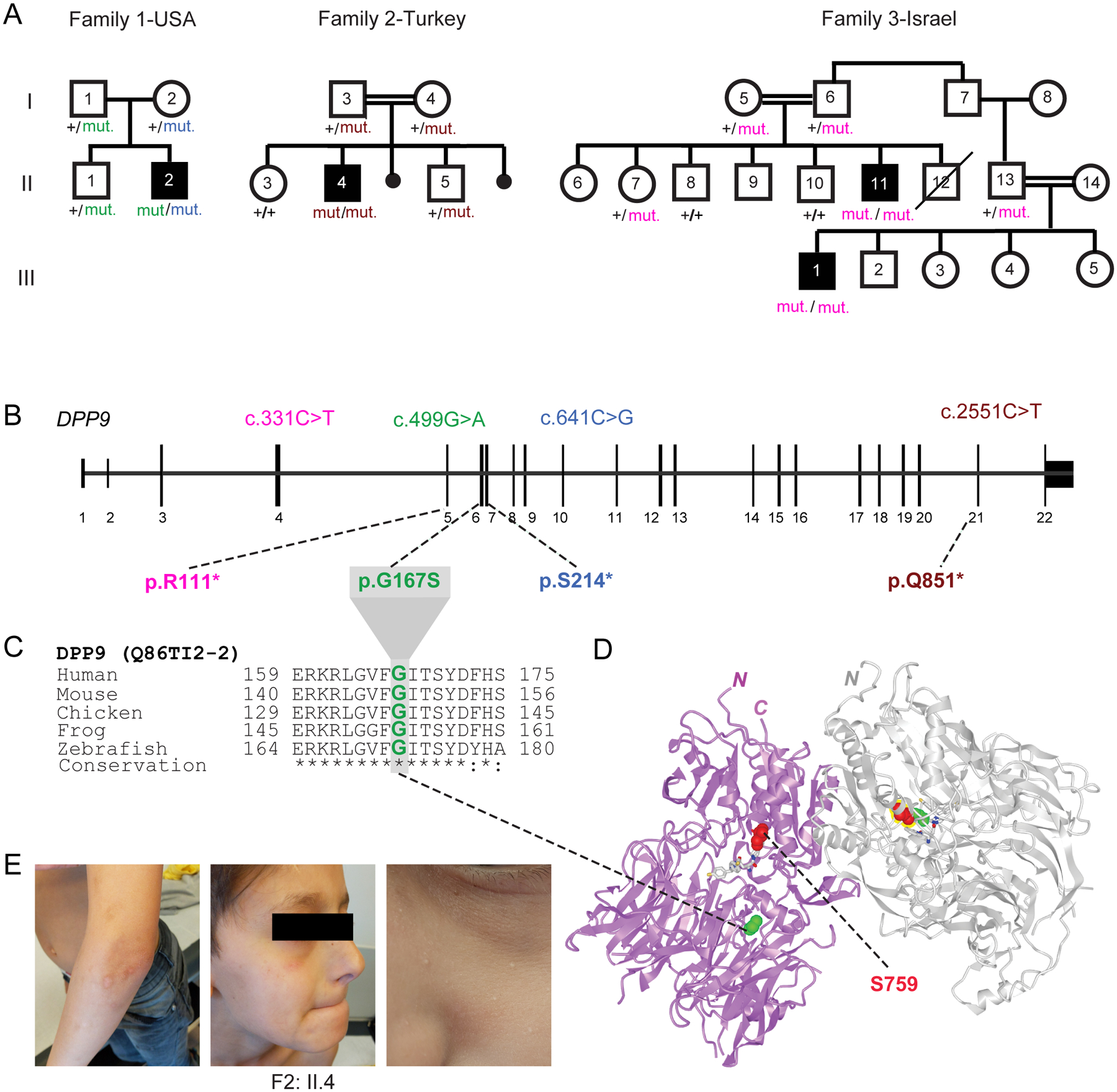
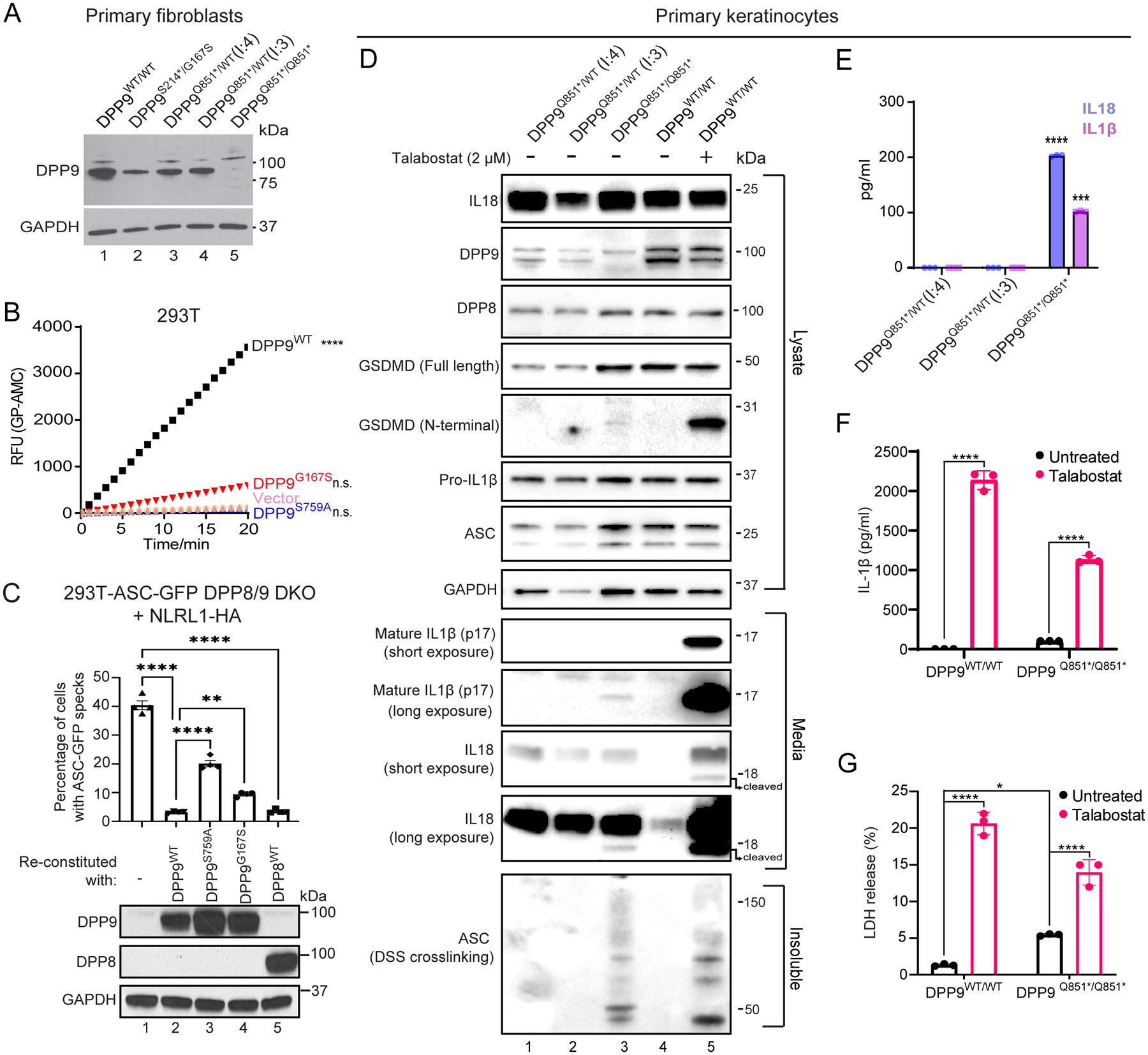
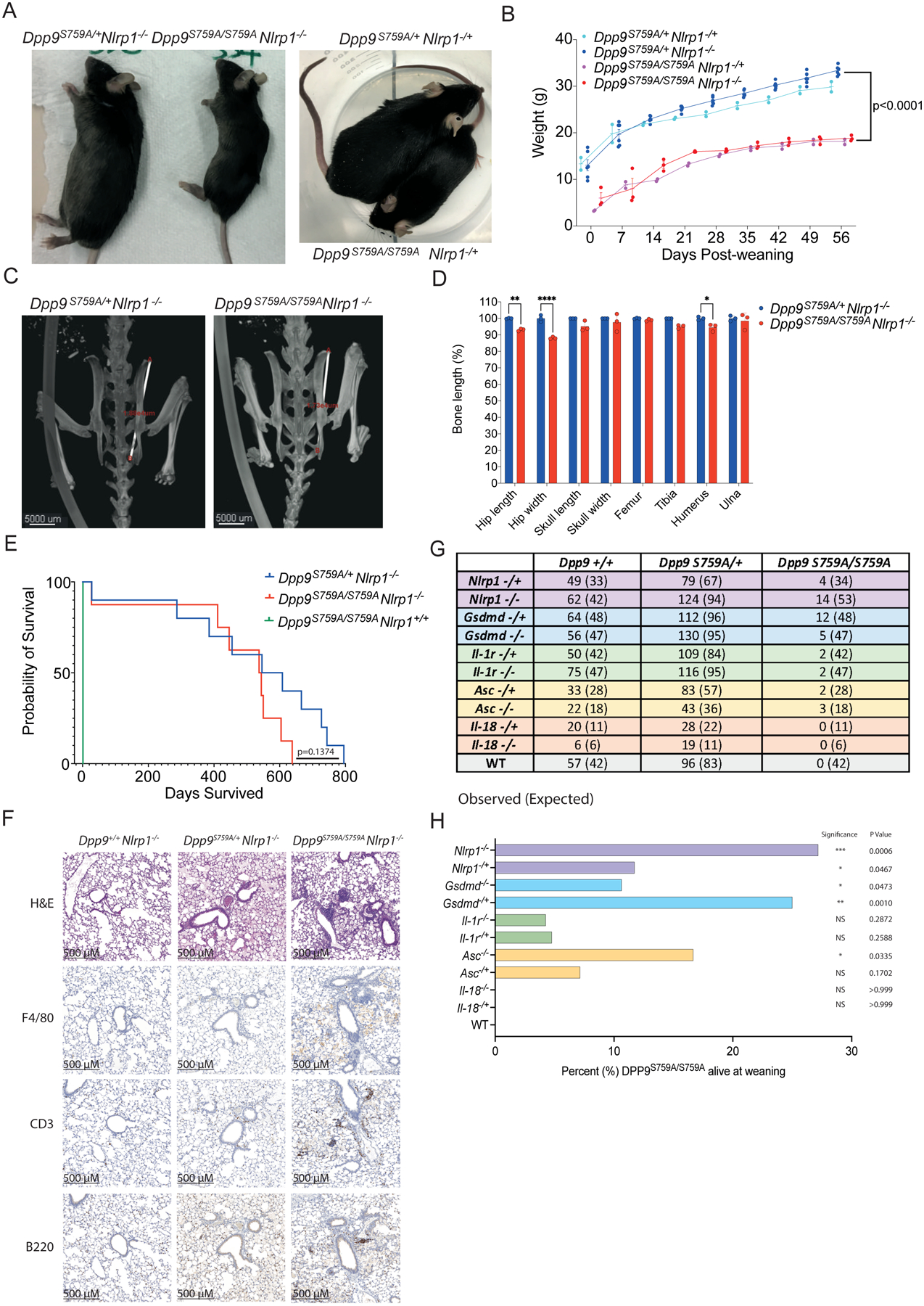
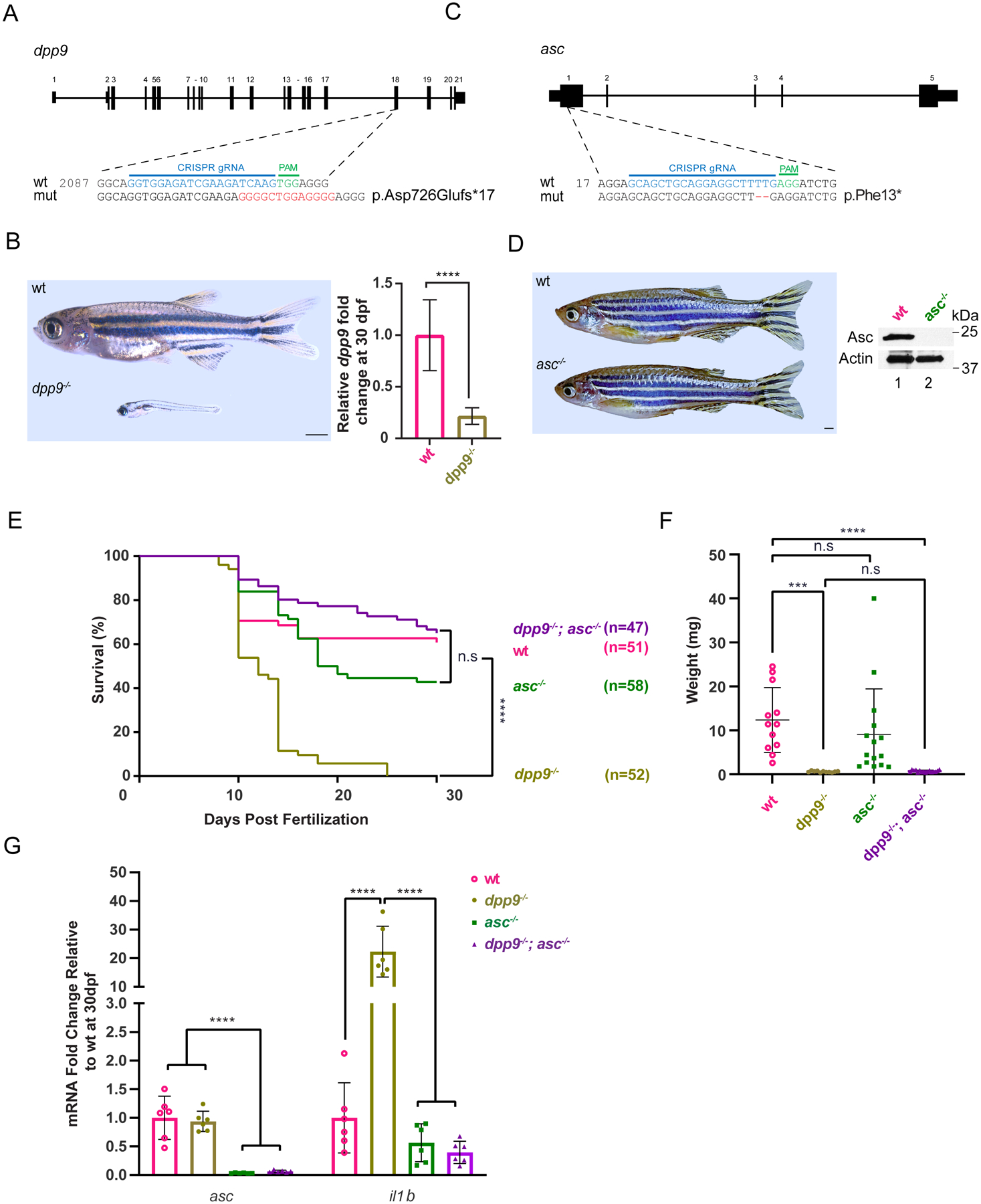
Dipeptidyl peptidase 9 (DPP9) is a direct inhibitor of NLRP1, but how it affects inflammasome regulation in vivo is not yet established. Here, we report three families with immune-associated defects, poor growth, pancytopenia, and skin pigmentation abnormalities that segregate with biallelic DPP9 rare variants. Using patient-derived primary cells and biochemical assays, these variants were shown to behave as hypomorphic or knockout alleles that failed to repress NLRP1. The removal of a single copy of Nlrp1a/b/c, Asc, Gsdmd, or Il-1r, but not Il-18, was sufficient to rescue the lethality of Dpp9 mutant neonates in mice. Similarly, dpp9 deficiency was partially rescued by the inactivation of asc, an obligate downstream adapter of the NLRP1 inflammasome, in zebrafish. These experiments suggest that the deleterious consequences of DPP9 deficiency were mostly driven by the aberrant activation of the canonical NLRP1 inflammasome and IL-1β signaling. Collectively, our results delineate a Mendelian disorder of DPP9 deficiency driven by increased NLRP1 activity as demonstrated in patient cells and in two animal models of the disease.
Conflict of interest statement
Competing interests:
S.L.M. is a Scientific Advisor for IFM Therapeutics and NRG Therapeutics.
Figures
References
-
- Fenini G, Grossi S, Contassot E, Biedermann T, Reichmann E, French LE, Beer H-D, Genome Editing of Human Primary Keratinocytes by CRISPR/Cas9 Reveals an Essential Role of the NLRP1 Inflammasome in UVB Sensing. J. Invest. Dermatol 138, 2644–2652 (2018). - PubMed
-
- Broz P, Dixit VM, Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol 16, 407–420 (2016). - PubMed
-
- Zhong FL, Mamaï O, Sborgi L, Boussofara L, Hopkins R, Robinson K, Szeverényi I, Takeichi T, Balaji R, Lau A, Tye H, Roy K, Bonnard C, Ahl PJ, Jones LA, Baker PJ, Lacina L, Otsuka A, Fournie PR, Malecaze F, Lane EB, Akiyama M, Kabashima K, Connolly JE, Masters SL, Soler VJ, Omar SS, McGrath JA, Nedelcu R, Gribaa M, Denguezli M, Saad A, Hiller S, Reversade B, Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell. 167, 187–202.e17 (2016). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous