

Mucopolysaccharidoses and the blood-brain barrier
- PMID: 36117162
- PMCID: PMC9484072
- DOI: 10.1186/s12987-022-00373-5
Mucopolysaccharidoses and the blood-brain barrier
Abstract
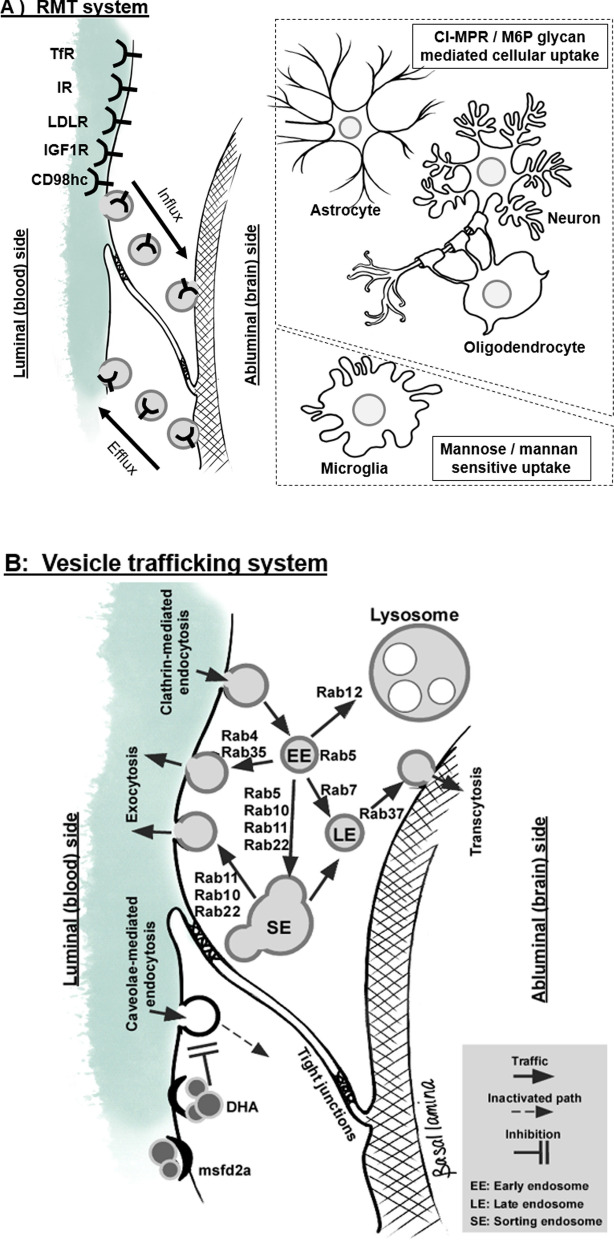
Mucopolysaccharidoses comprise a set of genetic diseases marked by an enzymatic dysfunction in the degradation of glycosaminoglycans in lysosomes. There are eight clinically distinct types of mucopolysaccharidosis, some with various subtypes, based on which lysosomal enzyme is deficient and symptom severity. Patients with mucopolysaccharidosis can present with a variety of symptoms, including cognitive dysfunction, hepatosplenomegaly, skeletal abnormalities, and cardiopulmonary issues. Additionally, the onset and severity of symptoms can vary depending on the specific disorder, with symptoms typically arising during early childhood. While there is currently no cure for mucopolysaccharidosis, there are clinically approved therapies for the management of clinical symptoms, such as enzyme replacement therapy. Enzyme replacement therapy is typically administered intravenously, which allows for the systemic delivery of the deficient enzymes to peripheral organ sites. However, crossing the blood-brain barrier (BBB) to ameliorate the neurological symptoms of mucopolysaccharidosis continues to remain a challenge for these large macromolecules. In this review, we discuss the transport mechanisms for the delivery of lysosomal enzymes across the BBB. Additionally, we discuss the several therapeutic approaches, both preclinical and clinical, for the treatment of mucopolysaccharidoses.
Keywords: Blood–brain barrier; Enzyme replacement therapy; Lysosomal storage disease; Mucopolysaccharidosis.
© 2022. The Author(s).
Conflict of interest statement
All authors have nothing to declare.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
