

The neuroprotective effects of glucagon-like peptide 1 in Alzheimer's and Parkinson's disease: An in-depth review
- PMID: 36117625
- PMCID: PMC9475012
- DOI: 10.3389/fnins.2022.970925
The neuroprotective effects of glucagon-like peptide 1 in Alzheimer's and Parkinson's disease: An in-depth review
Abstract
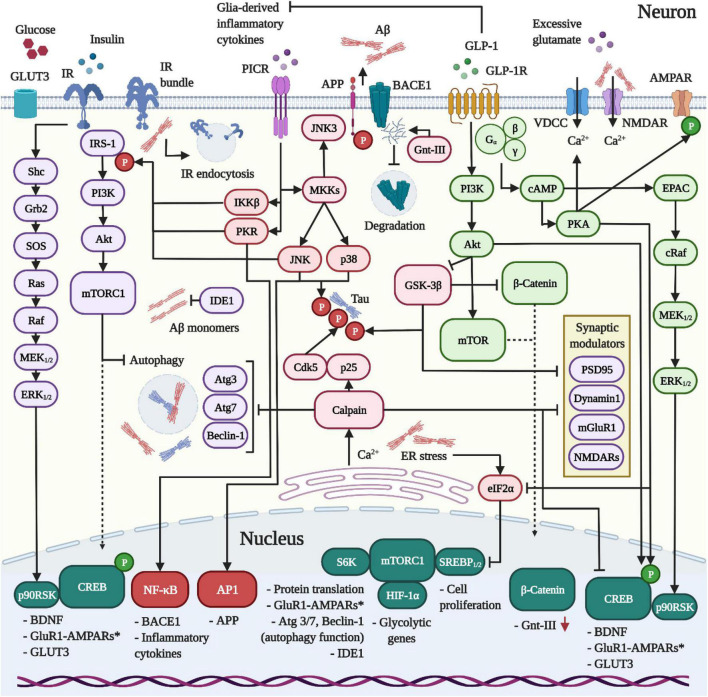
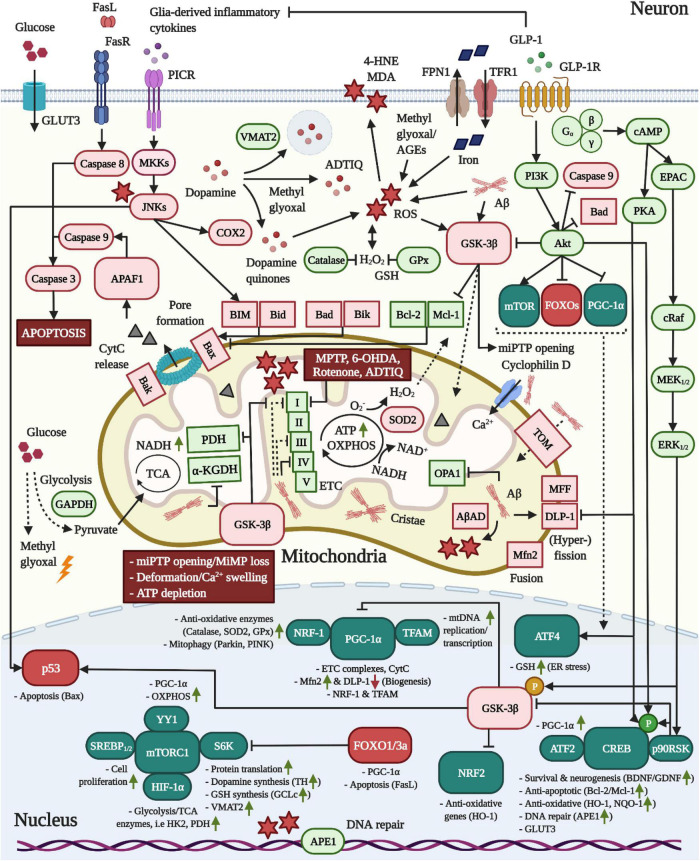
Currently, there is no disease-modifying treatment available for Alzheimer's and Parkinson's disease (AD and PD) and that includes the highly controversial approval of the Aβ-targeting antibody aducanumab for the treatment of AD. Hence, there is still an unmet need for a neuroprotective drug treatment in both AD and PD. Type 2 diabetes is a risk factor for both AD and PD. Glucagon-like peptide 1 (GLP-1) is a peptide hormone and growth factor that has shown neuroprotective effects in preclinical studies, and the success of GLP-1 mimetics in phase II clinical trials in AD and PD has raised new hope. GLP-1 mimetics are currently on the market as treatments for type 2 diabetes. GLP-1 analogs are safe, well tolerated, resistant to desensitization and well characterized in the clinic. Herein, we review the existing evidence and illustrate the neuroprotective pathways that are induced following GLP-1R activation in neurons, microglia and astrocytes. The latter include synaptic protection, improvements in cognition, learning and motor function, amyloid pathology-ameliorating properties (Aβ, Tau, and α-synuclein), the suppression of Ca2+ deregulation and ER stress, potent anti-inflammatory effects, the blockage of oxidative stress, mitochondrial dysfunction and apoptosis pathways, enhancements in the neuronal insulin sensitivity and energy metabolism, functional improvements in autophagy and mitophagy, elevated BDNF and glial cell line-derived neurotrophic factor (GDNF) synthesis as well as neurogenesis. The many beneficial features of GLP-1R and GLP-1/GIPR dual agonists encourage the development of novel drug treatments for AD and PD.
Keywords: Alzheimer’s disease; GLP-1; Parkinson’s disease; amyloid beta; brain glucose hypometabolism; insulin resistance; mitochondrial dysfunction; neuroinflammation.
Copyright © 2022 Reich and Hölscher.
Conflict of interest statement
CH was a named inventor on patents and patent applications that cover the use of GLP-1, GIP and dual GLP-1/GIP receptor agonists as treatments for neurodegenerative disorders. CH was also the CSO of the company Kariya Pharmaceuticals. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Therapeutic potentials of plant iridoids in Alzheimer's and Parkinson's diseases: A review.Eur J Med Chem. 2019 May 1;169:185-199. doi: 10.1016/j.ejmech.2019.03.009. Epub 2019 Mar 8. Eur J Med Chem. 2019. PMID: 30877973 Review.
-
Antidiabetic agents as a novel treatment for Alzheimer's and Parkinson's disease.Ageing Res Rev. 2023 Aug;89:101979. doi: 10.1016/j.arr.2023.101979. Epub 2023 Jun 14. Ageing Res Rev. 2023. PMID: 37328112 Review.
-
Glucagon-like peptide-1 (GLP-1) receptor agonists and neuroinflammation: Implications for neurodegenerative disease treatment.Pharmacol Res. 2022 Dec;186:106550. doi: 10.1016/j.phrs.2022.106550. Epub 2022 Nov 11. Pharmacol Res. 2022. PMID: 36372278 Free PMC article. Review.
-
Incretin-based drugs as potential therapy for neurodegenerative diseases: current status and perspectives.Pharmacol Ther. 2022 Nov;239:108277. doi: 10.1016/j.pharmthera.2022.108277. Epub 2022 Sep 3. Pharmacol Ther. 2022. PMID: 36064147 Review.
-
Beyond insulin: The Intriguing role of GLP-1 in Parkinson's disease.Eur J Pharmacol. 2024 Nov 5;982:176936. doi: 10.1016/j.ejphar.2024.176936. Epub 2024 Aug 23. Eur J Pharmacol. 2024. PMID: 39182542 Review.
Cited by
-
Comparing regional brain uptake of incretin receptor agonists after intranasal delivery in CD-1 mice and the APP/PS1 mouse model of Alzheimer's disease.Alzheimers Res Ther. 2024 Aug 1;16(1):173. doi: 10.1186/s13195-024-01537-1. Alzheimers Res Ther. 2024. PMID: 39085976 Free PMC article.
-
Immunoassay-based quantification of full-length peptidylglycine alpha-amidating monooxygenase in human plasma.Sci Rep. 2023 Jul 4;13(1):10827. doi: 10.1038/s41598-023-37976-3. Sci Rep. 2023. PMID: 37402878 Free PMC article.
-
Glucagon-like peptide-1 receptor agonists, but not dipeptidyl peptidase-4 inhibitors, reduce alcohol intake.J Clin Invest. 2025 Mar 6;135(9):e188314. doi: 10.1172/JCI188314. eCollection 2025 May 1. J Clin Invest. 2025. PMID: 40048376 Free PMC article.
-
Glucagon-Like Peptide 1 Therapy: From Discovery to Type 2 Diabetes and Beyond.Endocrinol Metab (Seoul). 2023 Feb;38(1):25-33. doi: 10.3803/EnM.2022.1642. Epub 2023 Feb 6. Endocrinol Metab (Seoul). 2023. PMID: 36740965 Free PMC article.
-
Microglial Drivers of Alzheimer's Disease Pathology: An Evolution of Diverse Participating States.Proteins. 2025 Aug;93(8):1330-1348. doi: 10.1002/prot.26723. Epub 2024 Sep 1. Proteins. 2025. PMID: 39219300 Free PMC article. Review.
References
-
- Agrawal R., Zhuang Y., Cummings B. P., Stanhope K. L., Graham J. L., Havel P. J., et al. (2014). Deterioration of plasticity and metabolic homeostasis in the brain of the UCD-T2DM rat model of naturally occurring type-2 diabetes. Biochim. Biophys. Acta 1842 1313–1323. 10.1016/j.bbadis.2014.05.007 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous