

Oxygen regulation of breathing is abolished in mitochondrial complex III-deficient arterial chemoreceptors
- PMID: 36122208
- PMCID: PMC9522341
- DOI: 10.1073/pnas.2202178119
Oxygen regulation of breathing is abolished in mitochondrial complex III-deficient arterial chemoreceptors
Abstract
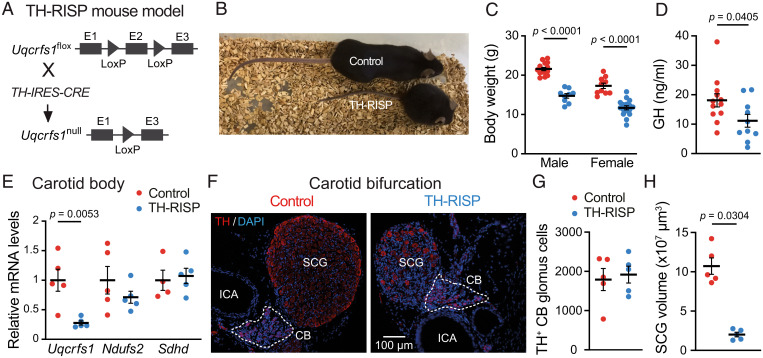
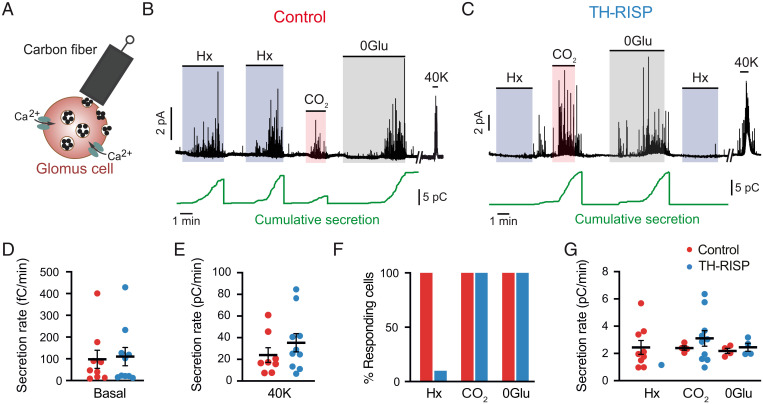
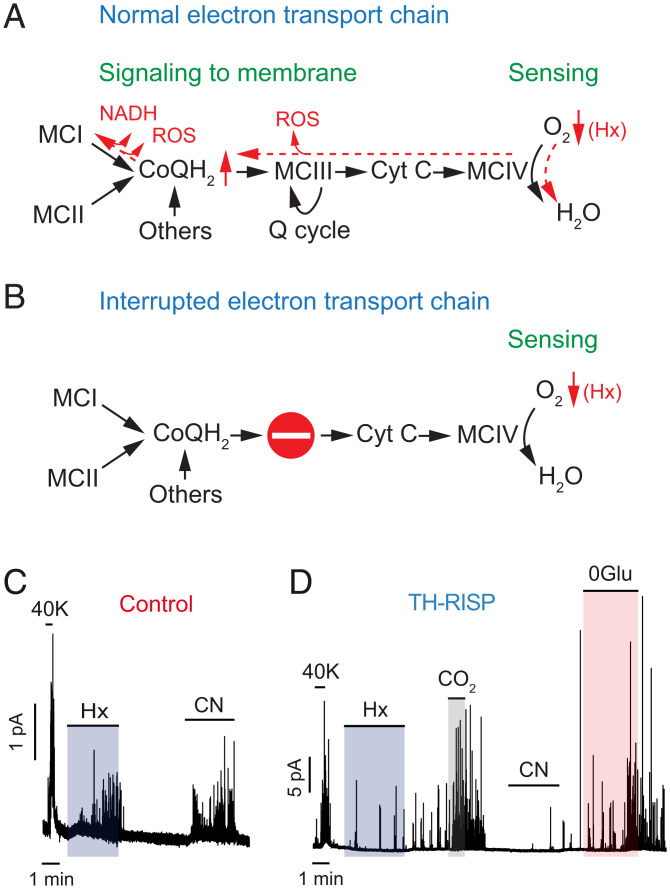
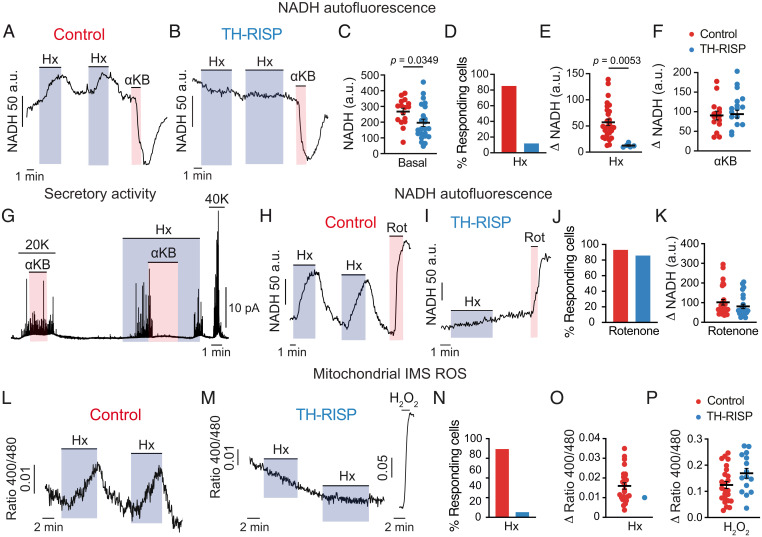
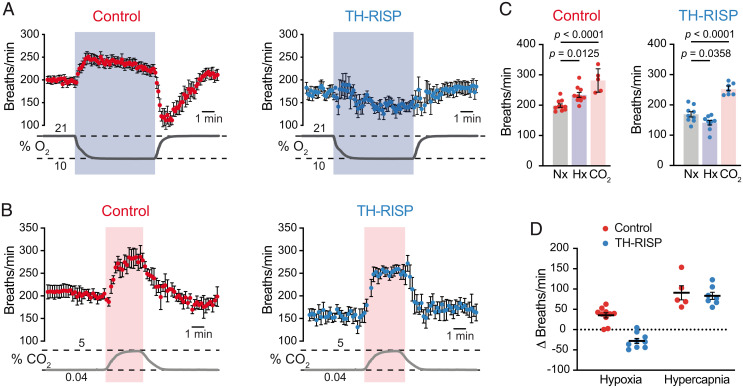
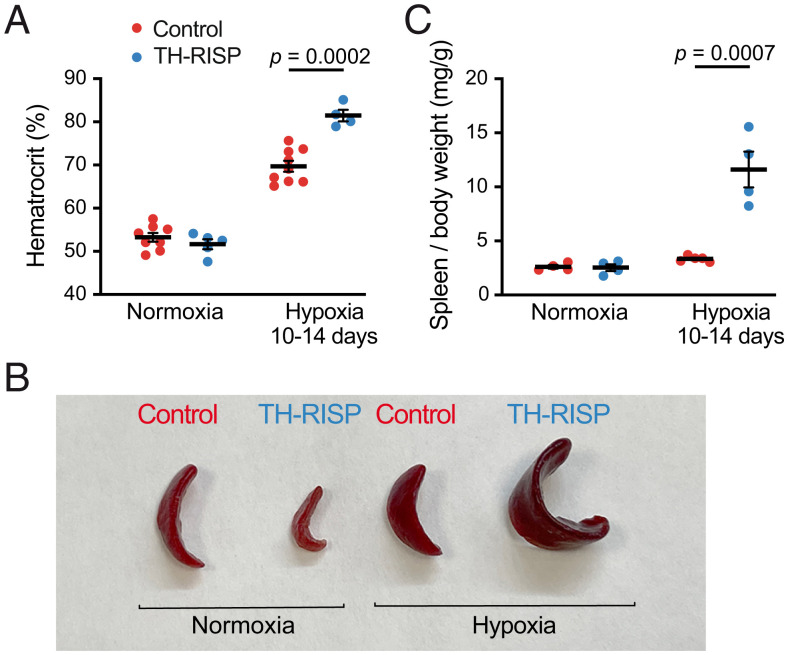
Acute oxygen (O2) sensing is essential for adaptation of organisms to hypoxic environments or medical conditions with restricted exchange of gases in the lung. The main acute O2-sensing organ is the carotid body (CB), which contains neurosecretory chemoreceptor (glomus) cells innervated by sensory fibers whose activation by hypoxia elicits hyperventilation and increased cardiac output. Glomus cells have mitochondria with specialized metabolic and electron transport chain (ETC) properties. Reduced mitochondrial complex (MC) IV activity by hypoxia leads to production of signaling molecules (NADH and reactive O2 species) in MCI and MCIII that modulate membrane ion channel activity. We studied mice with conditional genetic ablation of MCIII that disrupts the ETC in the CB and other catecholaminergic tissues. Glomus cells survived MCIII dysfunction but showed selective abolition of responsiveness to hypoxia (increased [Ca2+] and transmitter release) with normal responses to other stimuli. Mitochondrial hypoxic NADH and reactive O2 species signals were also suppressed. MCIII-deficient mice exhibited strong inhibition of the hypoxic ventilatory response and altered acclimatization to sustained hypoxia. These data indicate that a functional ETC, with coupling between MCI and MCIV, is required for acute O2 sensing. O2 regulation of breathing results from the integrated action of mitochondrial ETC complexes in arterial chemoreceptors.
Keywords: acute O2 sensing; carotid body glomus cell; hypoxia; mitochondrial O2 sensing and signaling; mitochondrial complex III.
Conflict of interest statement
The authors declare no competing interest.
Figures
Similar articles
-
Intracellular signalling in arterial chemoreceptors during acute hypoxia and glucose deprivation: role of ATP.J Physiol. 2025 Mar;603(5):1091-1107. doi: 10.1113/JP287130. Epub 2025 Feb 12. J Physiol. 2025. PMID: 39937745
-
Gene expression analyses reveal metabolic specifications in acute O2 -sensing chemoreceptor cells.J Physiol. 2017 Sep 15;595(18):6091-6120. doi: 10.1113/JP274684. Epub 2017 Aug 8. J Physiol. 2017. PMID: 28718507 Free PMC article.
-
Acute O2 sensing through HIF2α-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors.Sci Signal. 2020 Jan 21;13(615):eaay9452. doi: 10.1126/scisignal.aay9452. Sci Signal. 2020. PMID: 31848220
-
Redox signaling in acute oxygen sensing.Redox Biol. 2017 Aug;12:908-915. doi: 10.1016/j.redox.2017.04.033. Epub 2017 Apr 26. Redox Biol. 2017. PMID: 28476010 Free PMC article. Review.
-
Mitochondrial acute oxygen sensing and signaling.Crit Rev Biochem Mol Biol. 2022 Apr;57(2):205-225. doi: 10.1080/10409238.2021.2004575. Epub 2021 Dec 1. Crit Rev Biochem Mol Biol. 2022. PMID: 34852688 Review.
Cited by
-
Hif1α-dependent mitochondrial acute O2 sensing and signaling to myocyte Ca2+ channels mediate arterial hypoxic vasodilation.Nat Commun. 2024 Aug 5;15(1):6649. doi: 10.1038/s41467-024-51023-3. Nat Commun. 2024. PMID: 39103356 Free PMC article.
-
Mechanisms of Chemosensory Transduction in the Carotid Body.Adv Anat Embryol Cell Biol. 2023;237:49-62. doi: 10.1007/978-3-031-44757-0_5. Adv Anat Embryol Cell Biol. 2023. PMID: 37946077
-
The Oxygen Cascade from Atmosphere to Mitochondria as a Tool to Understand the (Mal)adaptation to Hypoxia.Int J Mol Sci. 2023 Feb 12;24(4):3670. doi: 10.3390/ijms24043670. Int J Mol Sci. 2023. PMID: 36835089 Free PMC article. Review.
-
A strategy for liver selective NRF2 induction via cytochrome P450-activated prodrugs with low activity in hypoxia.J Biol Chem. 2025 Jun;301(6):108487. doi: 10.1016/j.jbc.2025.108487. Epub 2025 Apr 8. J Biol Chem. 2025. PMID: 40209947 Free PMC article.
-
Transgenic NADH dehydrogenase restores oxygen regulation of breathing in mitochondrial complex I-deficient mice.Nat Commun. 2023 Mar 1;14(1):1172. doi: 10.1038/s41467-023-36894-2. Nat Commun. 2023. PMID: 36859533 Free PMC article.
References
-
- Ortega-Sáenz P., López-Barneo J., Physiology of the carotid body: From molecules to disease. Annu. Rev. Physiol. 82, 127–149 (2020). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
