

Therapeutic strategies in ischemic cardiomyopathy: Focus on mitochondrial quality surveillance
- PMID: 36122552
- PMCID: PMC9490489
- DOI: 10.1016/j.ebiom.2022.104260
Therapeutic strategies in ischemic cardiomyopathy: Focus on mitochondrial quality surveillance
Abstract
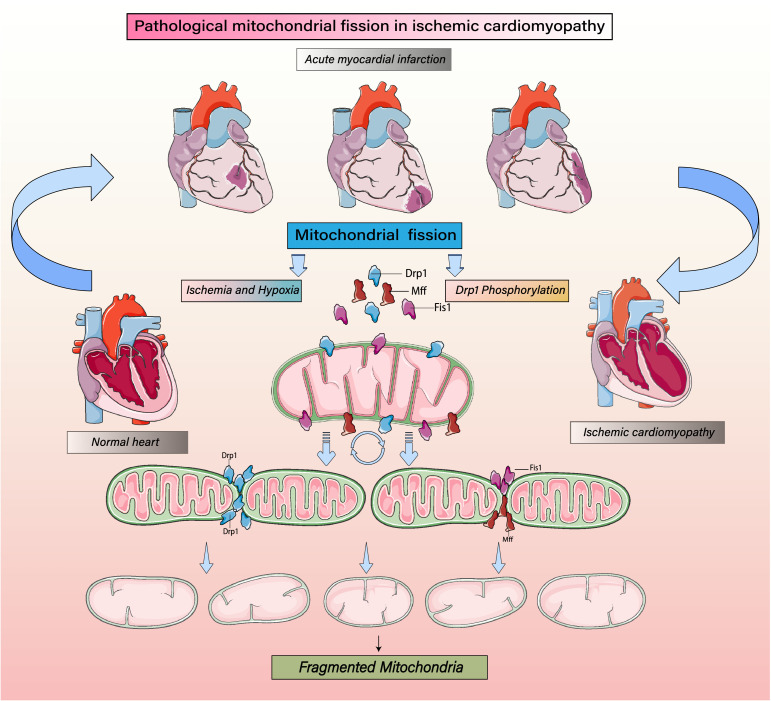
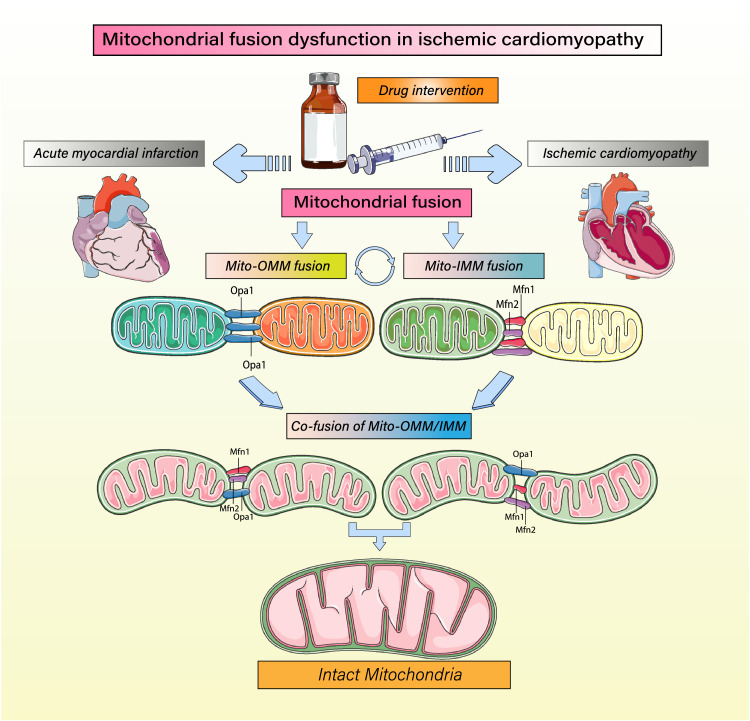
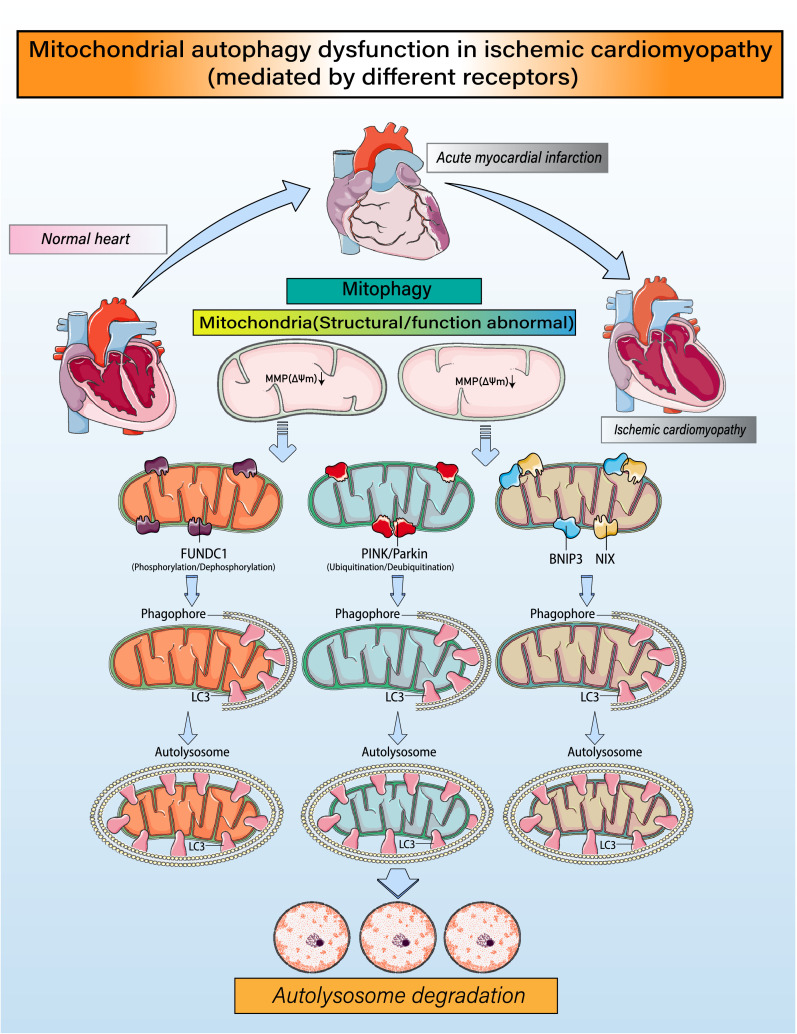
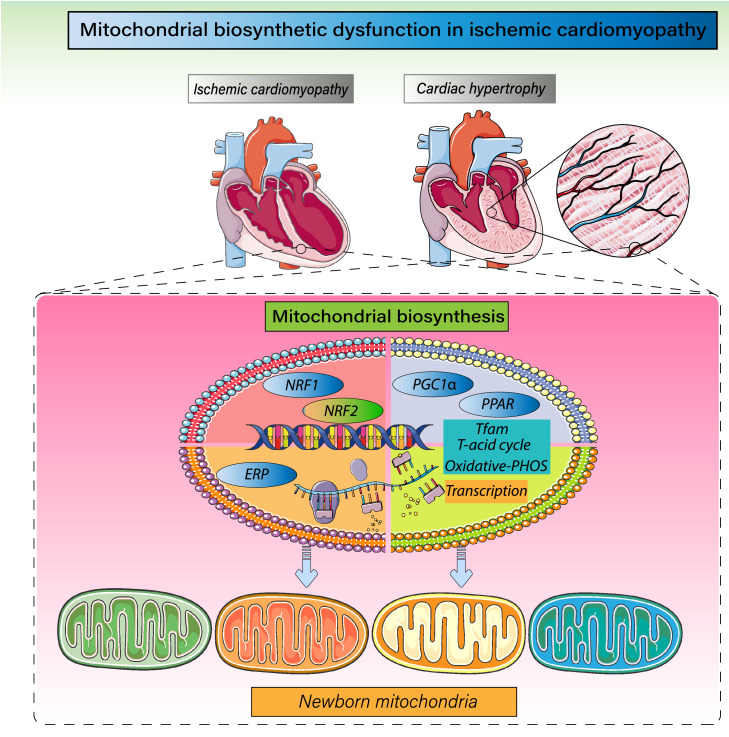
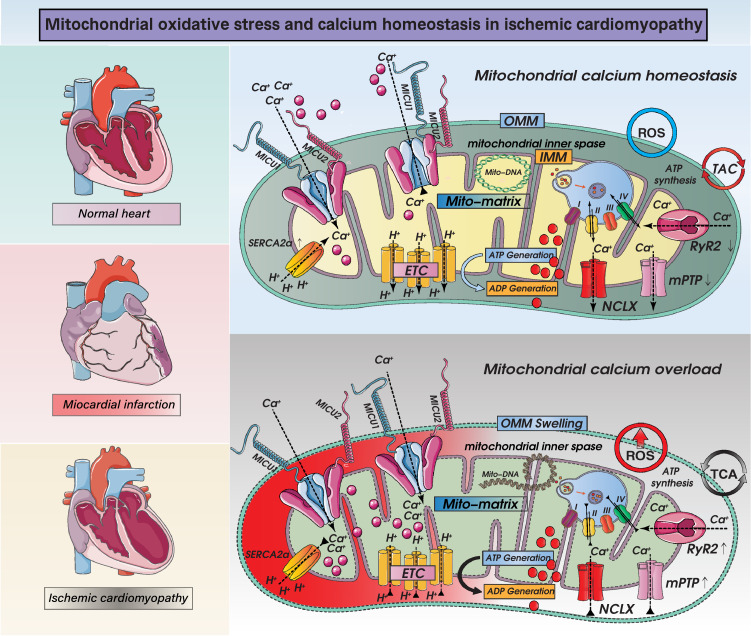
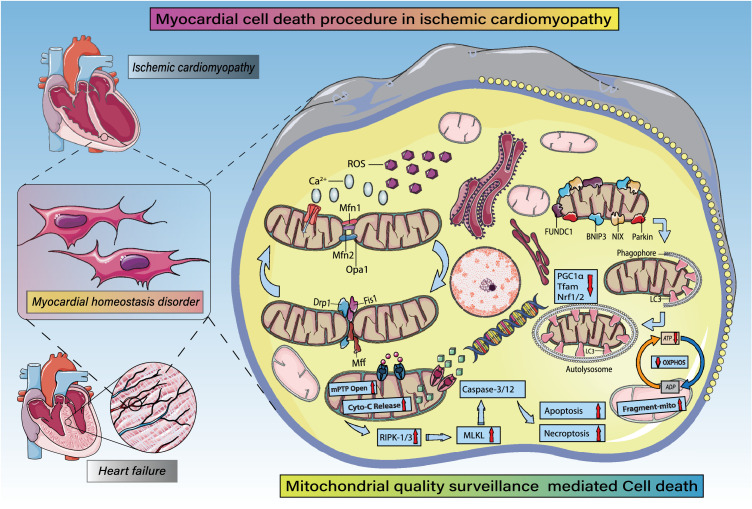
Despite considerable efforts to prevent and treat ischemic cardiomyopathy (ICM), effective therapies remain lacking, in part owing to the complexity of the underlying molecular mechanisms, which are not completely understood yet. It is now widely thought that mitochondria serve as "sentinel" organelles that are capable of detecting cellular injury and integrating multiple stress signals. These pathophysiological activities are temporally and spatially governed by the mitochondrial quality surveillance (MQS) system, involving mitochondrial dynamics, mitophagy, and biogenesis. Dysregulation of MQS is an early and critical process contributing to mitochondrial bioenergetic dysfunction and sublethal injury to cardiomyocytes during ICM. An improved understanding of the pathogenesis of ICM may enable the development of novel preventive and therapeutic strategies aimed at overcoming the challenge of myocardial ischemia and its cardiovascular sequelae. This review describes recent research on the protective effects of MQS in ICM and highlights promising therapeutic targets.
Keywords: Cardiomyocyte; Ischemic cardiomyopathy; Mitochondrial biogenesis; Mitochondrial dynamics; Mitochondrial quality surveillance; Mitophagy.
Copyright © 2022 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests The authors have no conflicts of interest to declare.
Figures
References
-
- Hearse DJ. Myocardial ischaemia: can we agree on a definition for the 21st century? Cardiovasc Res. 1994;28(12):1737–1744. discussion 45-6. - PubMed
-
- Cabac-Pogorevici I, Muk B, Rustamova Y, et al. Ischaemic cardiomyopathy. Pathophysiological insights, diagnostic management and the roles of revascularisation and device treatment. Gaps and dilemmas in the era of advanced technology. Eur J Heart Fail. 2020;22(5):789–799. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
