

The evolutionary dynamics of extrachromosomal DNA in human cancers
- PMID: 36123406
- PMCID: PMC9534767
- DOI: 10.1038/s41588-022-01177-x
The evolutionary dynamics of extrachromosomal DNA in human cancers
Abstract
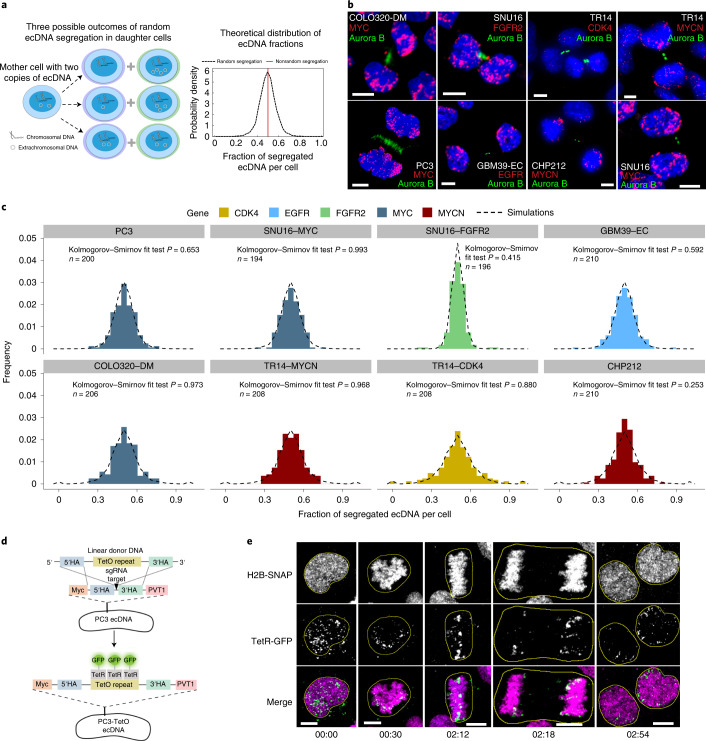
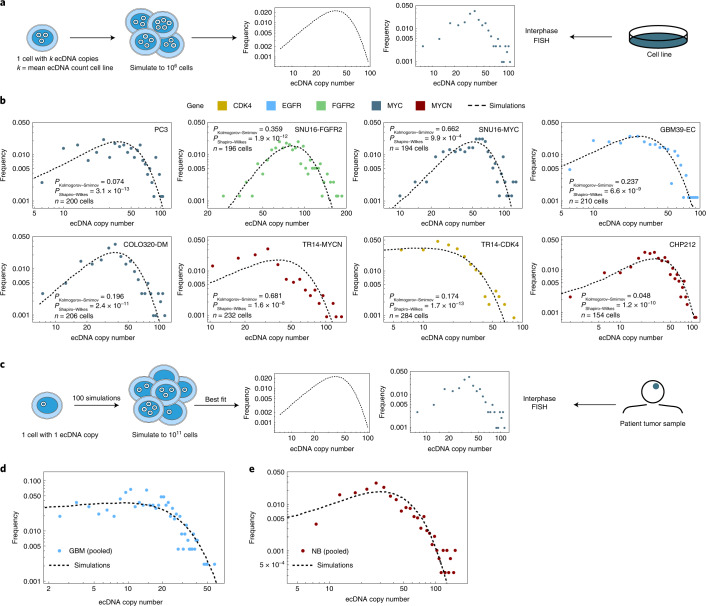
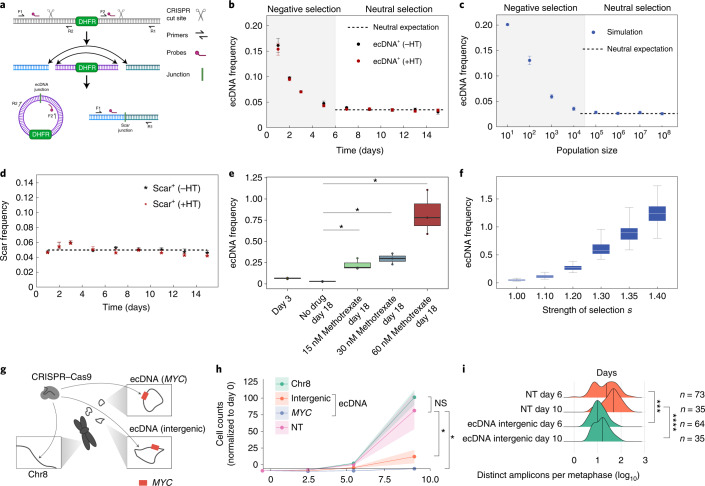
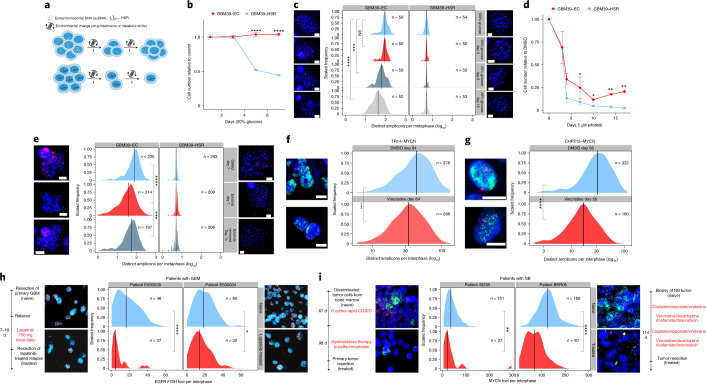
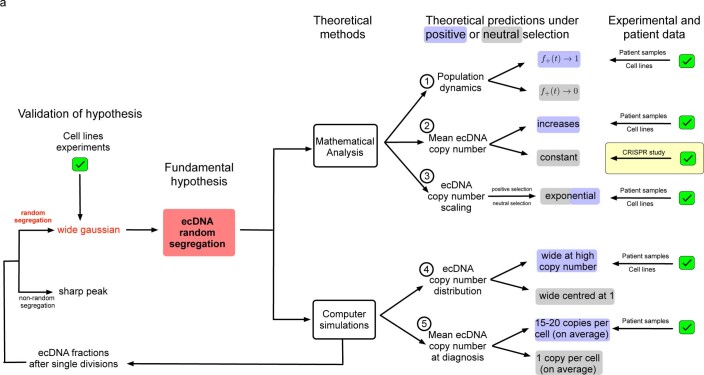
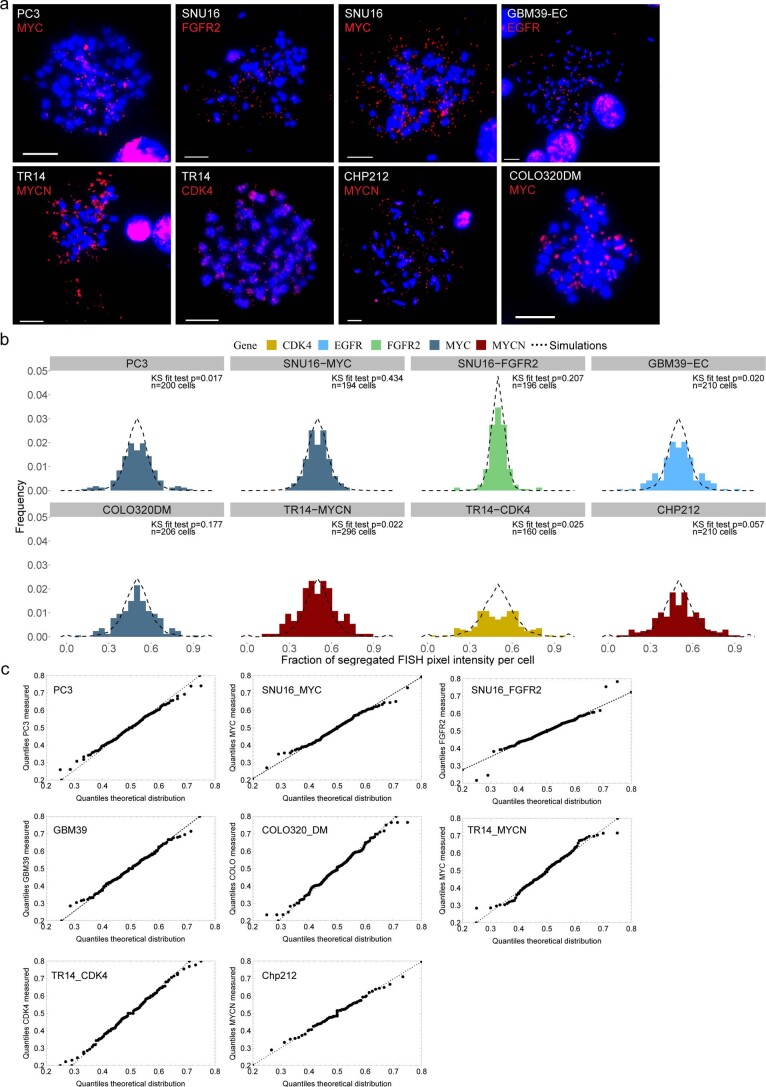
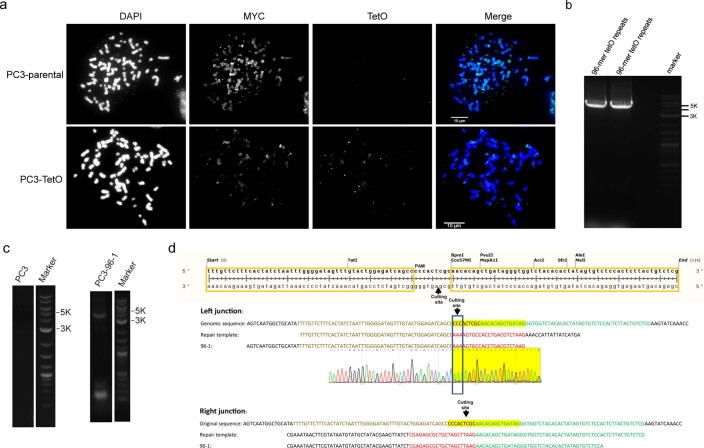
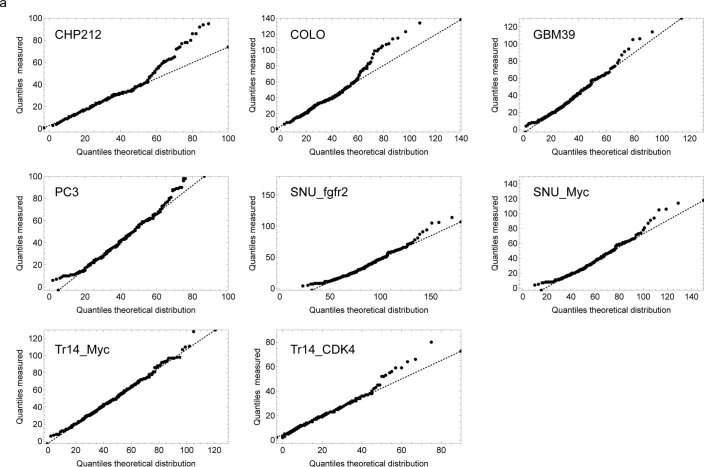
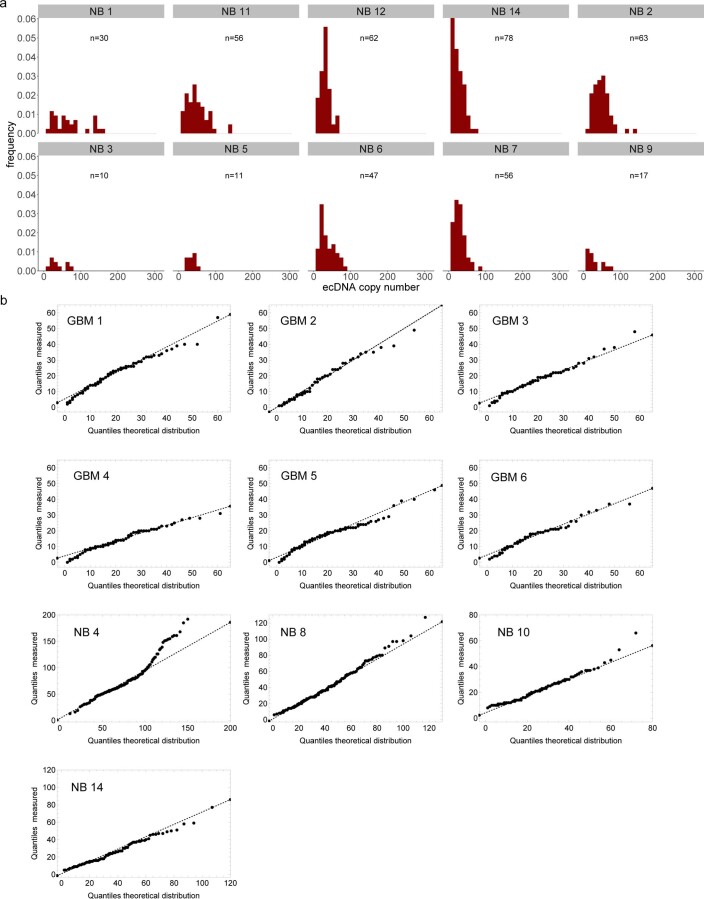
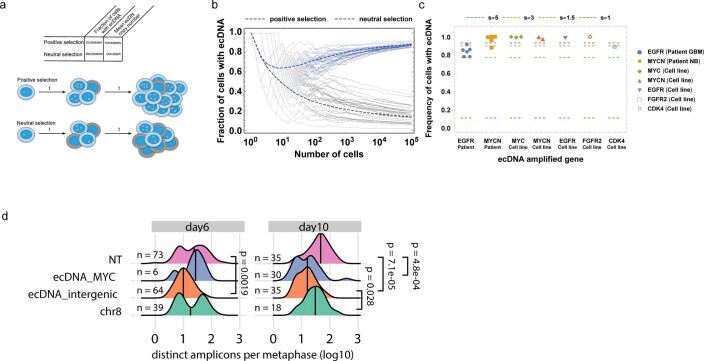
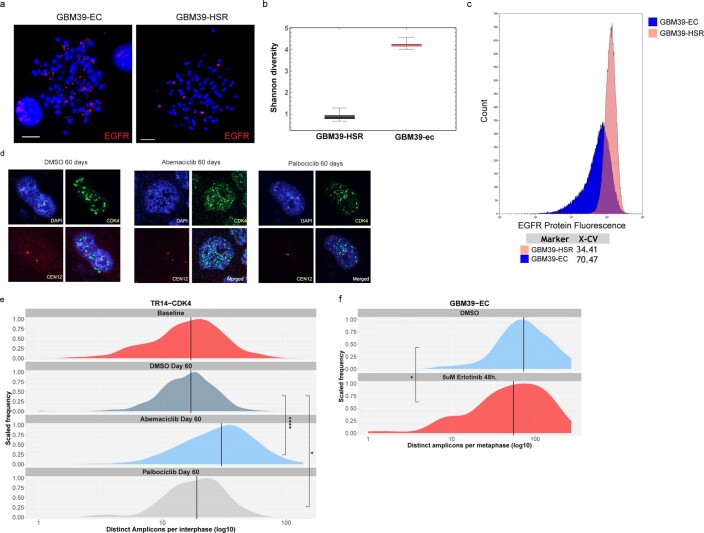
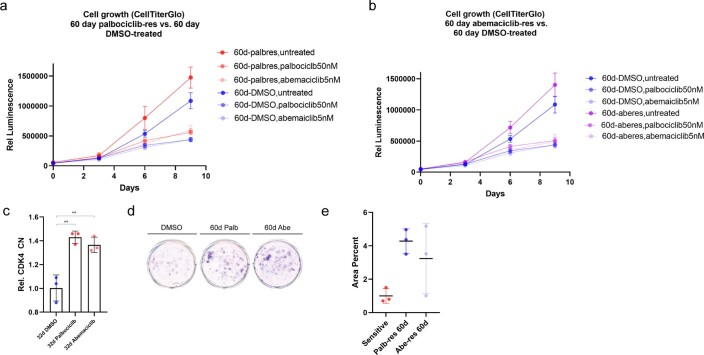
Oncogene amplification on extrachromosomal DNA (ecDNA) is a common event, driving aggressive tumor growth, drug resistance and shorter survival. Currently, the impact of nonchromosomal oncogene inheritance-random identity by descent-is poorly understood. Also unclear is the impact of ecDNA on somatic variation and selection. Here integrating theoretical models of random segregation, unbiased image analysis, CRISPR-based ecDNA tagging with live-cell imaging and CRISPR-C, we demonstrate that random ecDNA inheritance results in extensive intratumoral ecDNA copy number heterogeneity and rapid adaptation to metabolic stress and targeted treatment. Observed ecDNAs benefit host cell survival or growth and can change within a single cell cycle. ecDNA inheritance can predict, a priori, some of the aggressive features of ecDNA-containing cancers. These properties are facilitated by the ability of ecDNA to rapidly adapt genomes in a way that is not possible through chromosomal oncogene amplification. These results show how the nonchromosomal random inheritance pattern of ecDNA contributes to poor outcomes for patients with cancer.
© 2022. The Author(s).
Conflict of interest statement
P.S.M. is cofounder of Boundless Bio. He has equity and chairs the scientific advisory board, for which he is compensated. V.B. is a cofounder, consultant, scientific advisory board member and has an equity interest in Boundless Bio. and Abterra Biosciences. The terms of this arrangement have been reviewed and approved in accordance with its conflict-of-interest policies. H.Y.C. is a cofounder of Accent Therapeutics, Boundless Bio, Cartography Biosciences, Orbital Therapeutics, and advisor of 10X Genomics, Arsenal Biosciences and Spring Discovery. J.T.L. was employed by Boundless Bio after completing this work. C.S. acknowledges grant support from AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae (previously ArcherDX, collaboration in minimal residual disease sequencing technologies) and Ono Pharmaceutical. He is an AstraZeneca advisory board member and chief investigator for the AZ MeRmaiD 1 and 2 clinical trials and is also chief investigator of the NHS-Galleri trial. He has consulted for Amgen, AstraZeneca, Pfizer, Novartis, GSK, MSD, Bristol Myers Squibb, Illumina, Genentech, Roche-Ventana, GRAIL, Medicxi, Metabomed, Bicycle Therapeutics, Roche Innovation Centre Shanghai and the Sarah Cannon Research Institute. C.S. had stock options in ApoGen Biotechnologies and GRAIL until June 2021, has currently stock options in Epic Bioscience and Bicycle Therapeutics, and has stock options and is a cofounder of Achilles Therapeutics. C.S. holds patents relating to assay technology to detect tumor recurrence (PCT/GB2017/053289), targeting neoantigens (PCT/EP2016/059401), identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), determining human leukocyte antigen loss of heterozygosity (PCT/GB2018/052004), predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912), a US patent relating to detecting tumor mutations (PCT/US2017/28013), methods for lung cancer detection (US20190106751A1) and both European and US patents related to identifying insertion/deletion mutation targets (PCT/GB2018/051892). The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- P 35841/FWF_/Austrian Science Fund FWF/Austria
- R01 CA238249/CA/NCI NIH HHS/United States
- C11496/A17786/CRUK_/Cancer Research UK/United Kingdom
- F99 CA274692/CA/NCI NIH HHS/United States
- U24 CA264379/CA/NCI NIH HHS/United States
- FC001169/CRUK_/Cancer Research UK/United Kingdom
- C416/A21999/CRUK_/Cancer Research UK/United Kingdom
- I 4162/FWF_/Austrian Science Fund FWF/Austria
- R01 GM114362/GM/NIGMS NIH HHS/United States
- R35 CA209919/CA/NCI NIH HHS/United States
- C11496/A30025/CRUK_/Cancer Research UK/United Kingdom
- HHMI/Howard Hughes Medical Institute/United States
- DH_/Department of Health/United Kingdom
- CGCATF-2021/100012/CRUK_/Cancer Research UK/United Kingdom
- FC001169/MRC_/Medical Research Council/United Kingdom
- OT2 CA278635/CA/NCI NIH HHS/United States
- FC001169 /WT_/Wellcome Trust/United Kingdom
