

H3.3-G34 mutations impair DNA repair and promote cGAS/STING-mediated immune responses in pediatric high-grade glioma models
- PMID: 36125896
- PMCID: PMC9663161
- DOI: 10.1172/JCI154229
H3.3-G34 mutations impair DNA repair and promote cGAS/STING-mediated immune responses in pediatric high-grade glioma models
Abstract
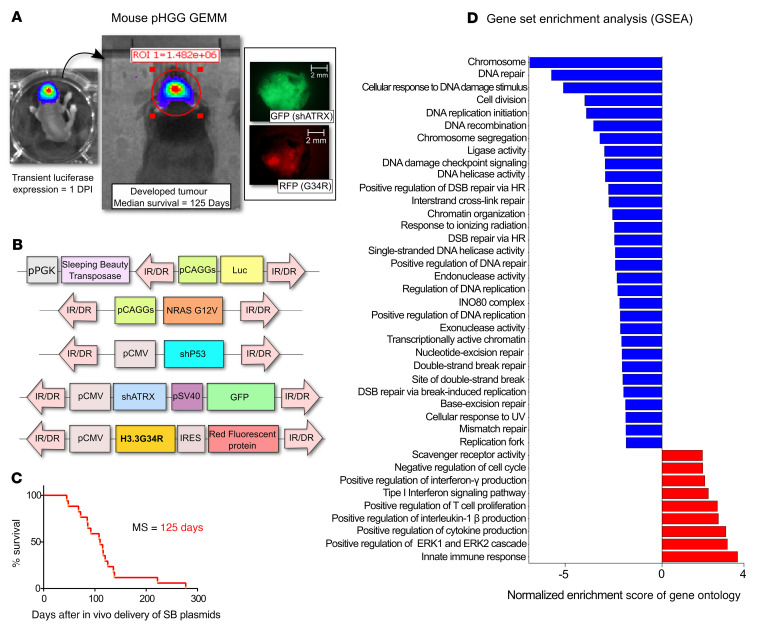
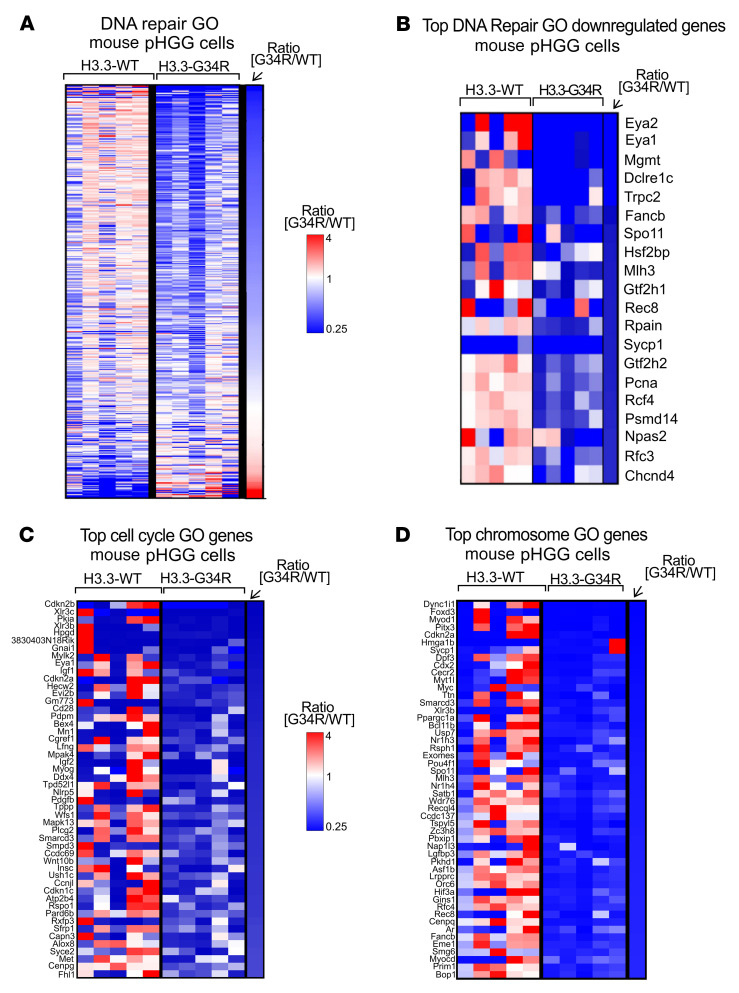
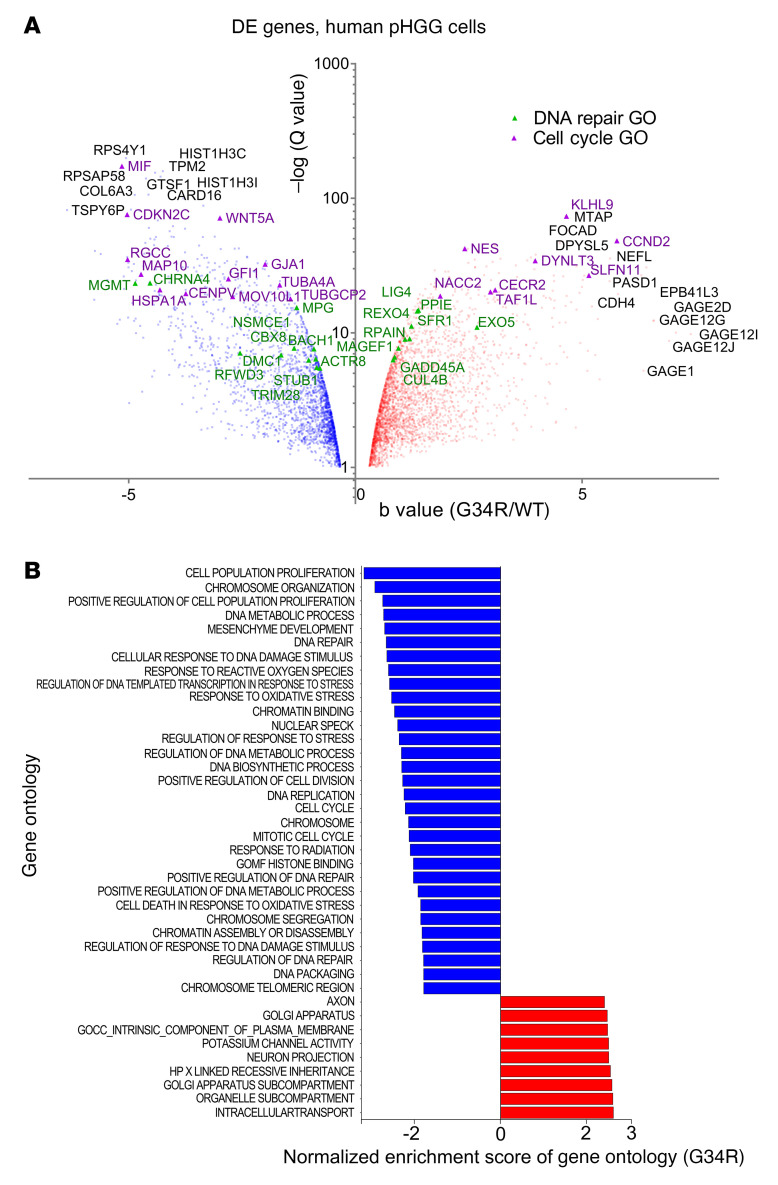
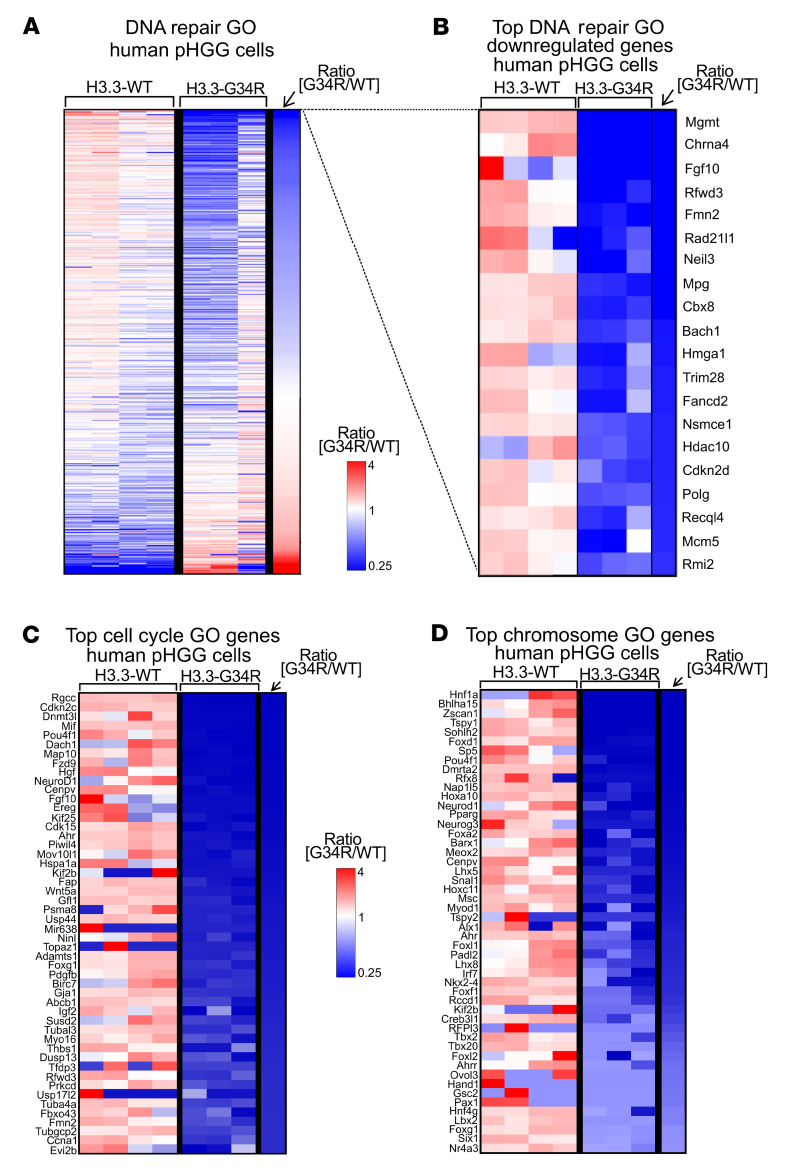
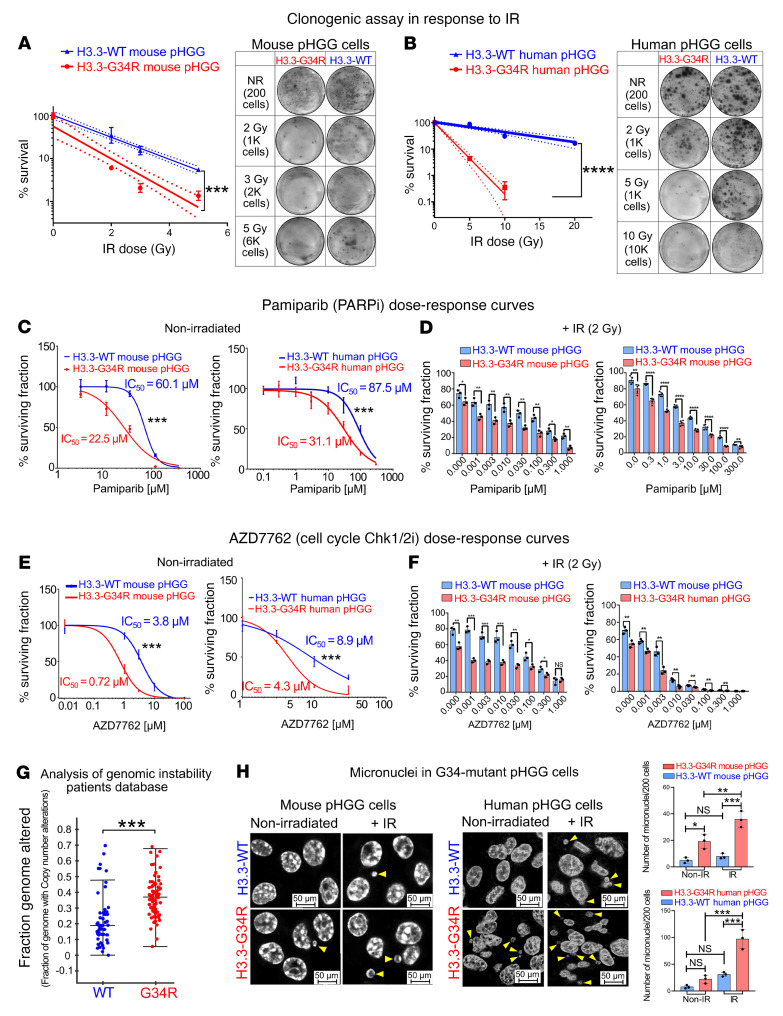
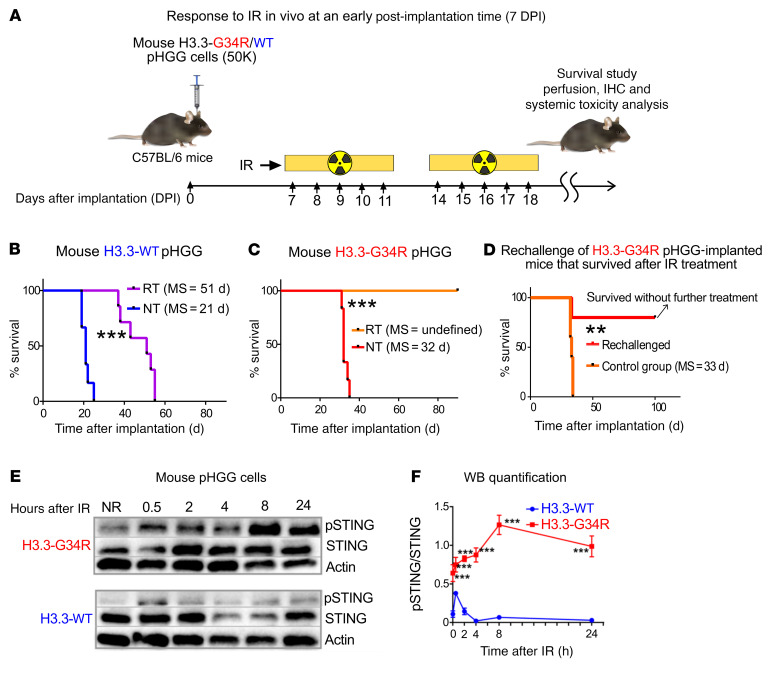
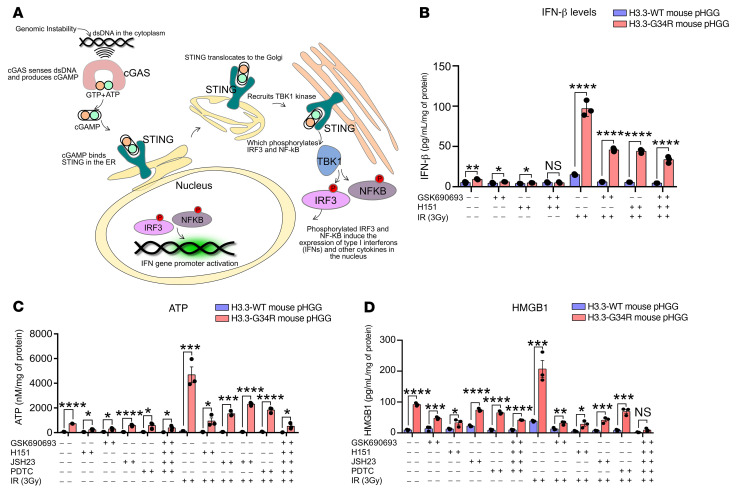
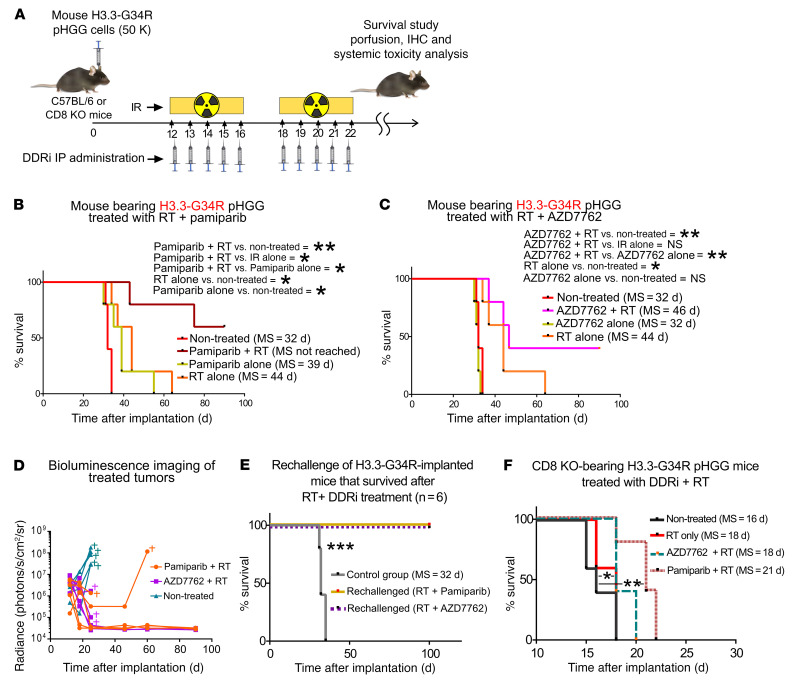
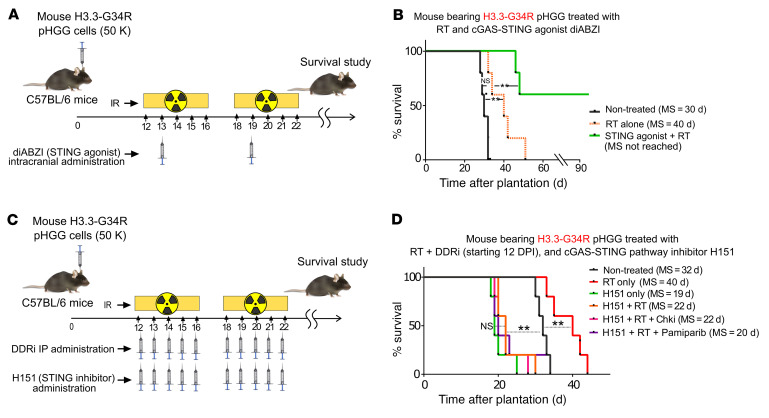
Pediatric high-grade gliomas (pHGGs) are the leading cause of cancer-related deaths in children in the USA. Sixteen percent of hemispheric pediatric and young adult HGGs encode Gly34Arg/Val substitutions in the histone H3.3 (H3.3-G34R/V). The mechanisms by which H3.3-G34R/V drive malignancy and therapeutic resistance in pHGGs remain unknown. Using a syngeneic, genetically engineered mouse model (GEMM) and human pHGG cells encoding H3.3-G34R, we demonstrate that this mutation led to the downregulation of DNA repair pathways. This resulted in enhanced susceptibility to DNA damage and inhibition of the DNA damage response (DDR). We demonstrate that genetic instability resulting from improper DNA repair in G34R-mutant pHGG led to the accumulation of extrachromosomal DNA, which activated the cyclic GMP-AMP synthase/stimulator of IFN genes (cGAS/STING) pathway, inducing the release of immune-stimulatory cytokines. We treated H3.3-G34R pHGG-bearing mice with a combination of radiotherapy (RT) and DNA damage response inhibitors (DDRi) (i.e., the blood-brain barrier-permeable PARP inhibitor pamiparib and the cell-cycle checkpoint CHK1/2 inhibitor AZD7762), and these combinations resulted in long-term survival for approximately 50% of the mice. Moreover, the addition of a STING agonist (diABZl) enhanced the therapeutic efficacy of these treatments. Long-term survivors developed immunological memory, preventing pHGG growth upon rechallenge. These results demonstrate that DDRi and STING agonists in combination with RT induced immune-mediated therapeutic efficacy in G34-mutant pHGG.
Keywords: Brain cancer; DNA repair; Drug therapy; Oncology; Therapeutics.
Conflict of interest statement
Figures
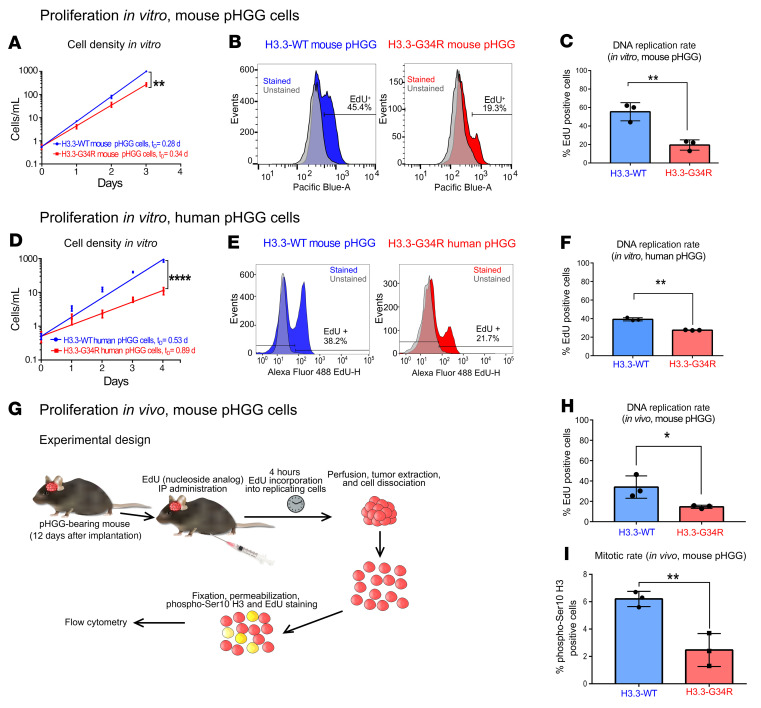
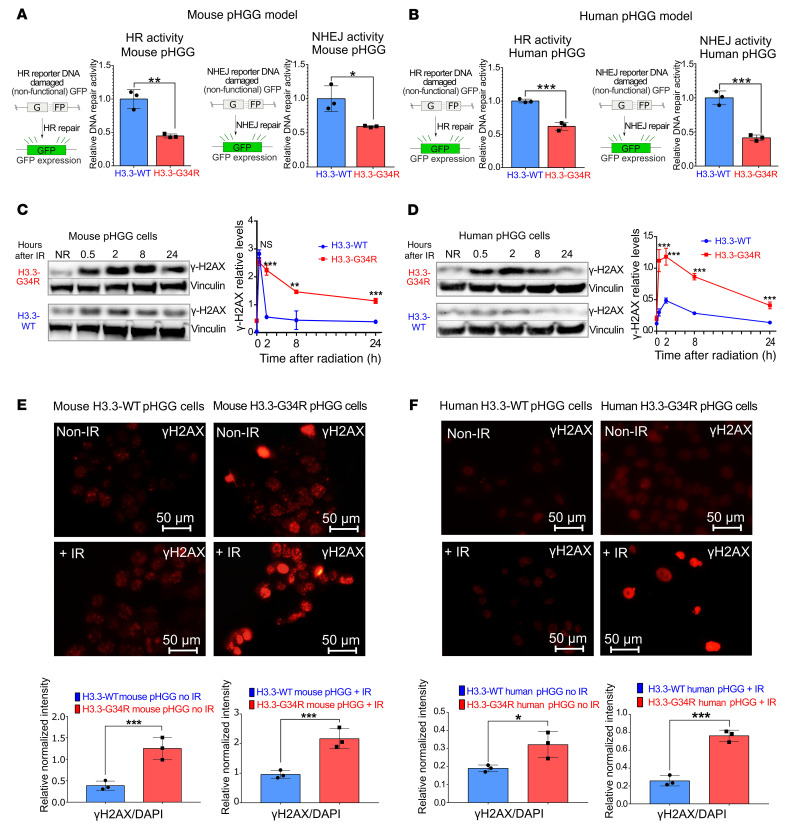
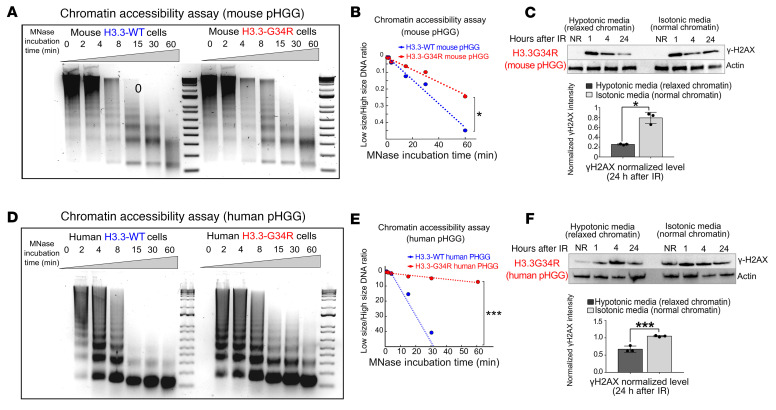
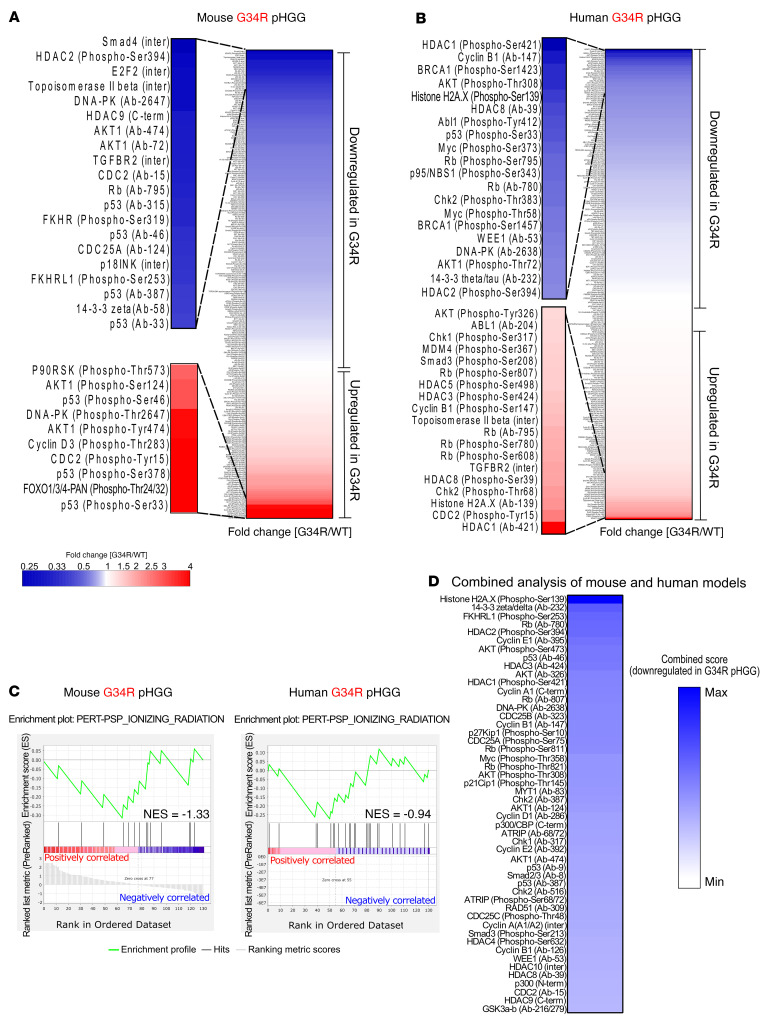
Comment in
-
STINGing the immune system: lessons learned through a model of G34-mutant pediatric high-grade glioma.J Clin Invest. 2022 Nov 15;132(22):e164420. doi: 10.1172/JCI164420. J Clin Invest. 2022. PMID: 36377657 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- R21 NS123879/NS/NINDS NIH HHS/United States
- R01 NS105556/NS/NINDS NIH HHS/United States
- R01 NS082311/NS/NINDS NIH HHS/United States
- R01 CA243916/CA/NCI NIH HHS/United States
- R01 NS076991/NS/NINDS NIH HHS/United States
- R37 NS094804/NS/NINDS NIH HHS/United States
- R01 NS124607/NS/NINDS NIH HHS/United States
- R01 NS096756/NS/NINDS NIH HHS/United States
- R01 NS122234/NS/NINDS NIH HHS/United States
- R01 NS122165/NS/NINDS NIH HHS/United States
- R01 NS122536/NS/NINDS NIH HHS/United States
- R01 NS110572/NS/NINDS NIH HHS/United States
- R01 NS124167/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
