

Exploring the mobilome and resistome of Enterococcus faecium in a One Health context across two continents
- PMID: 36129737
- PMCID: PMC9676038
- DOI: 10.1099/mgen.0.000880
Exploring the mobilome and resistome of Enterococcus faecium in a One Health context across two continents
Abstract
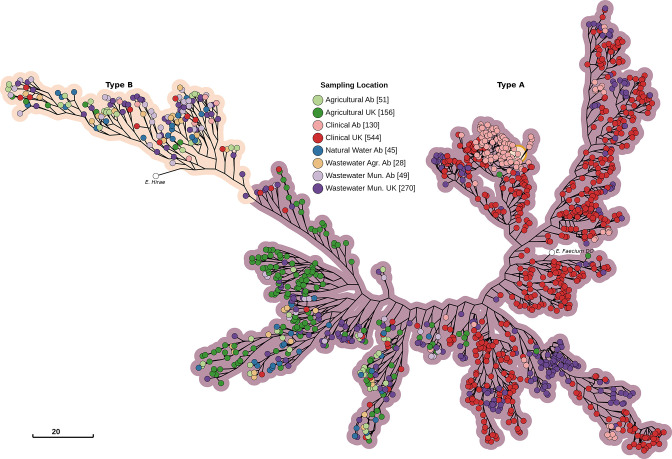
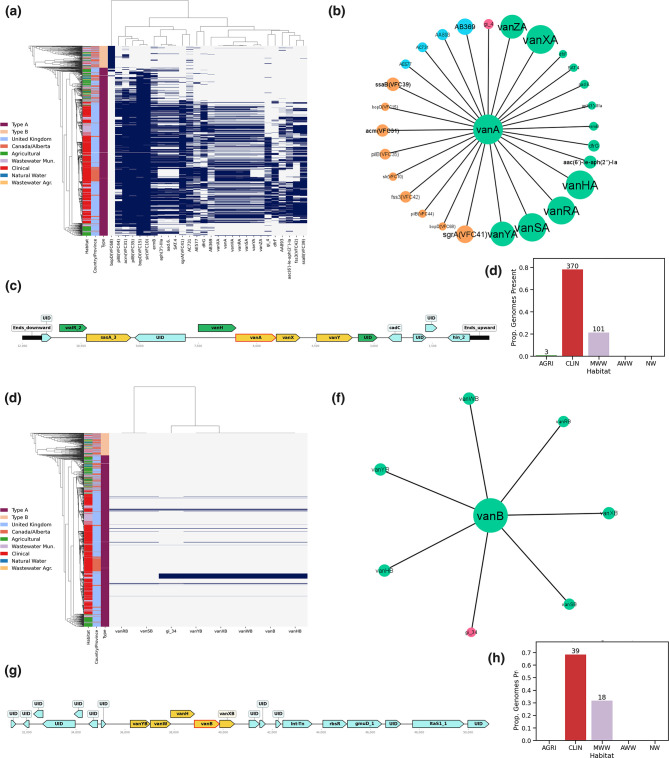
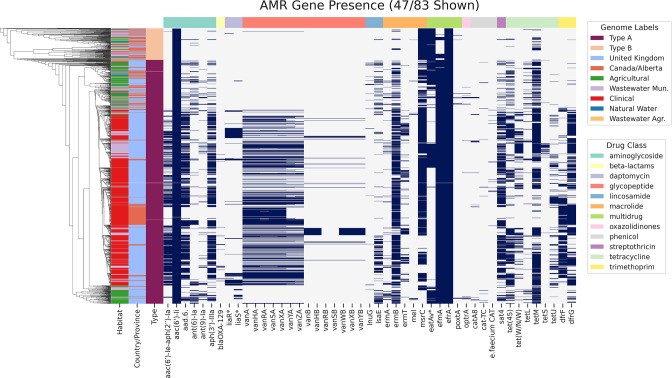
Enterococcus faecium is a ubiquitous opportunistic pathogen that is exhibiting increasing levels of antimicrobial resistance (AMR). Many of the genes that confer resistance and pathogenic functions are localized on mobile genetic elements (MGEs), which facilitate their transfer between lineages. Here, features including resistance determinants, virulence factors and MGEs were profiled in a set of 1273 E. faecium genomes from two disparate geographic locations (in the UK and Canada) from a range of agricultural, clinical and associated habitats. Neither lineages of E. faecium, type A and B, nor MGEs are constrained by geographic proximity, but our results show evidence of a strong association of many profiled genes and MGEs with habitat. Many features were associated with a group of clinical and municipal wastewater genomes that are likely forming a new human-associated ecotype within type A. The evolutionary dynamics of E. faecium make it a highly versatile emerging pathogen, and its ability to acquire, transmit and lose features presents a high risk for the emergence of new pathogenic variants and novel resistance combinations. This study provides a workflow for MGE-centric surveillance of AMR in Enterococcus that can be adapted to other pathogens.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
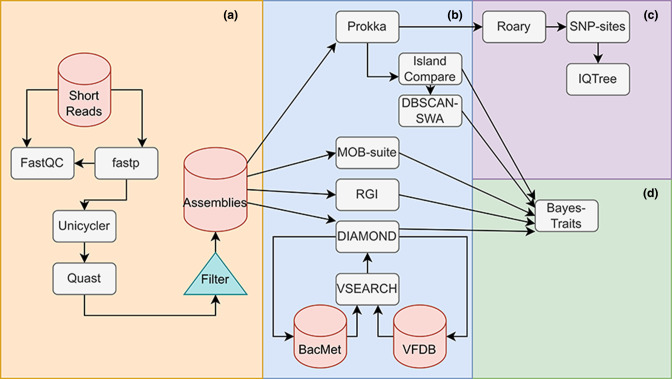
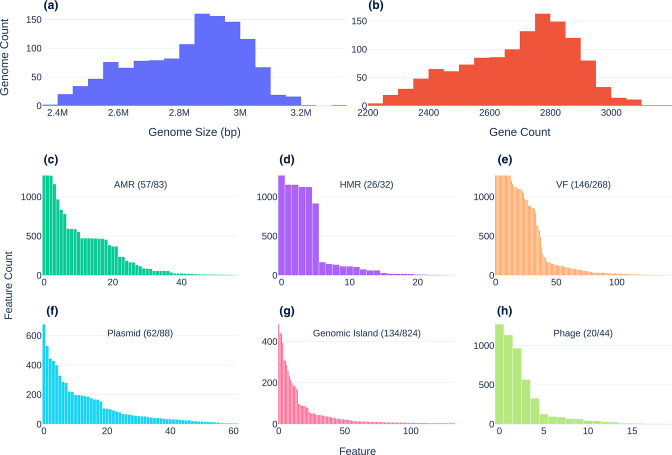
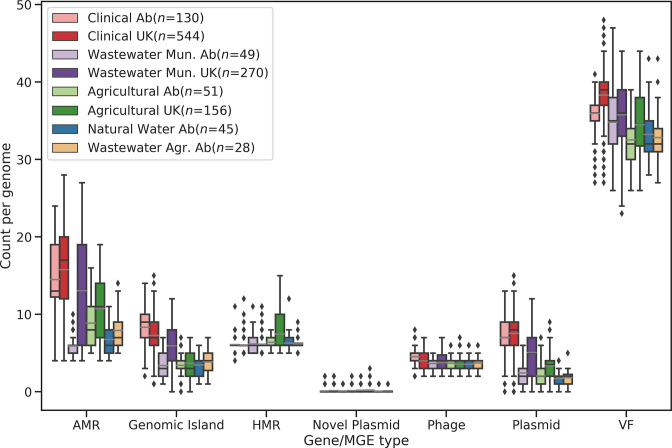
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources