

Genome-wide fetalization of enhancer architecture in heart disease
- PMID: 36130500
- PMCID: PMC9534044
- DOI: 10.1016/j.celrep.2022.111400
Genome-wide fetalization of enhancer architecture in heart disease
Abstract
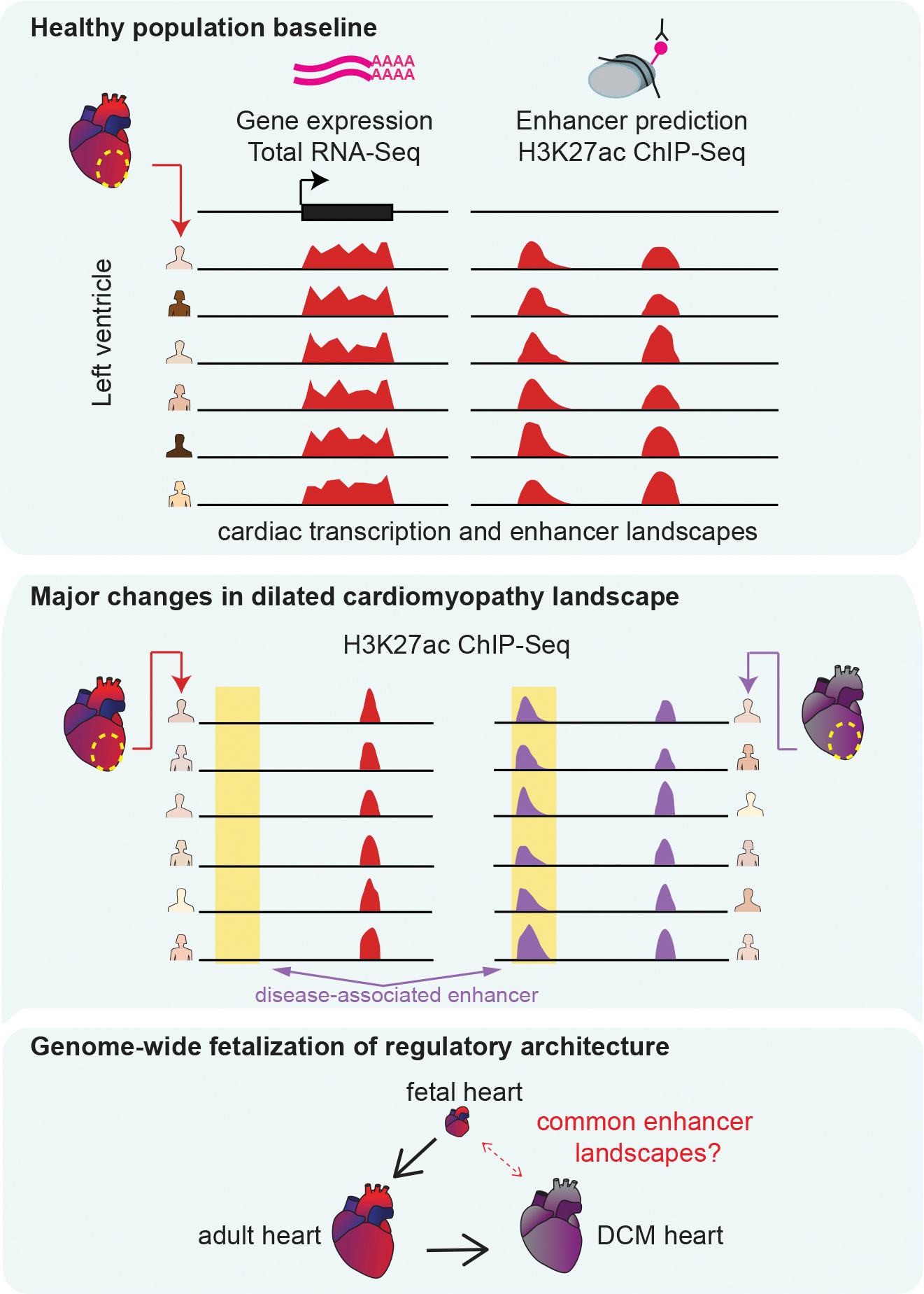
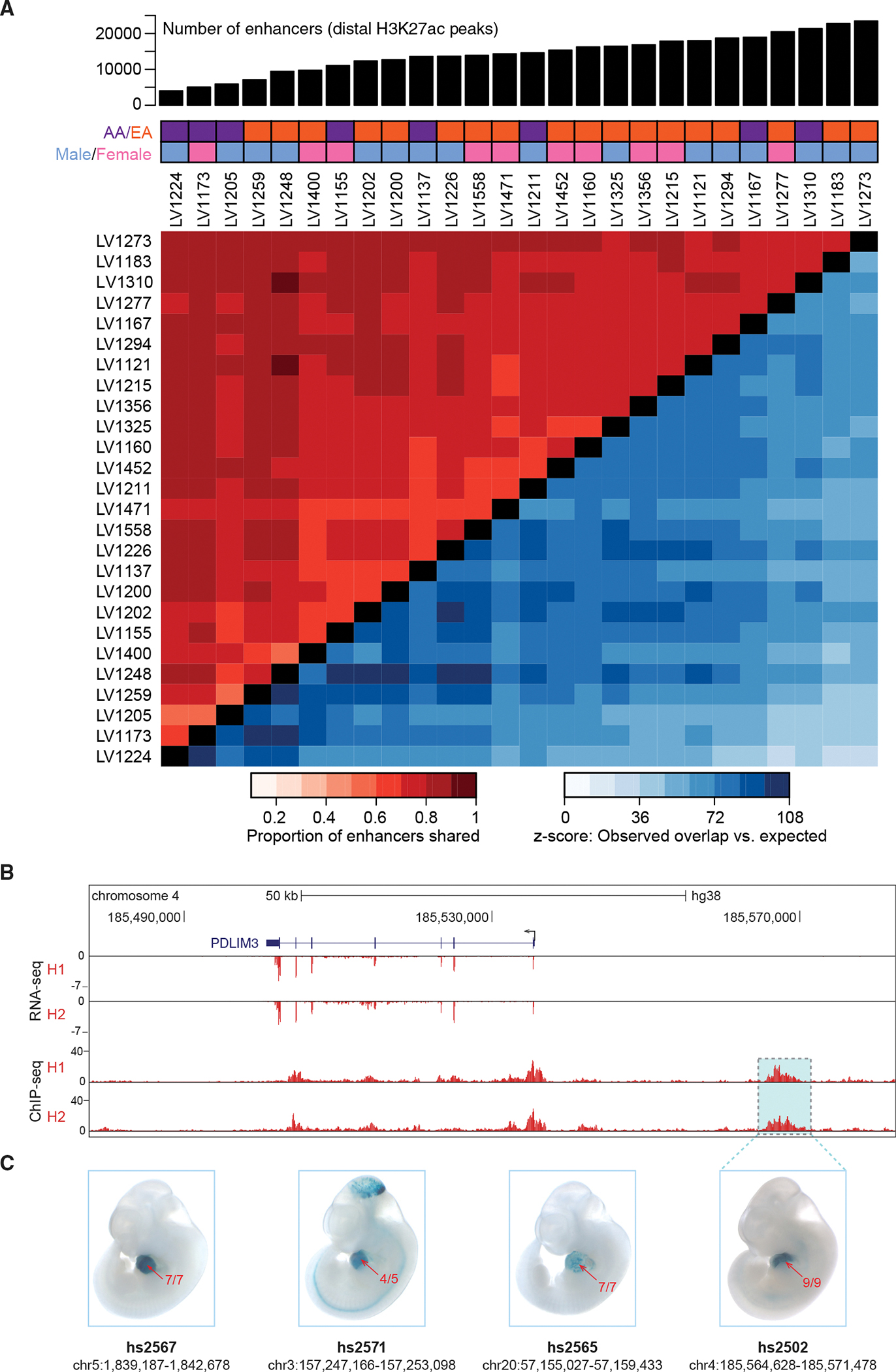
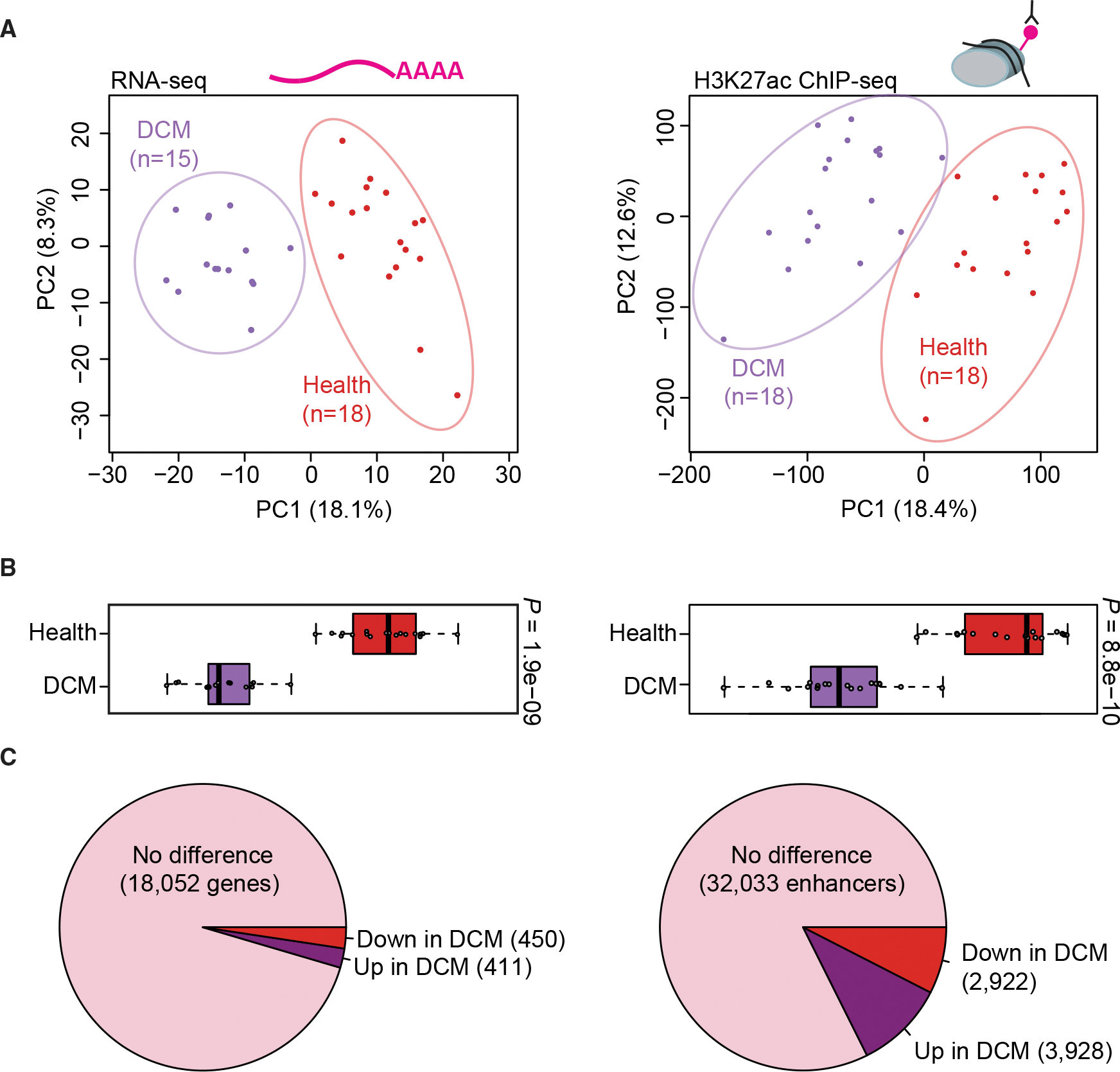
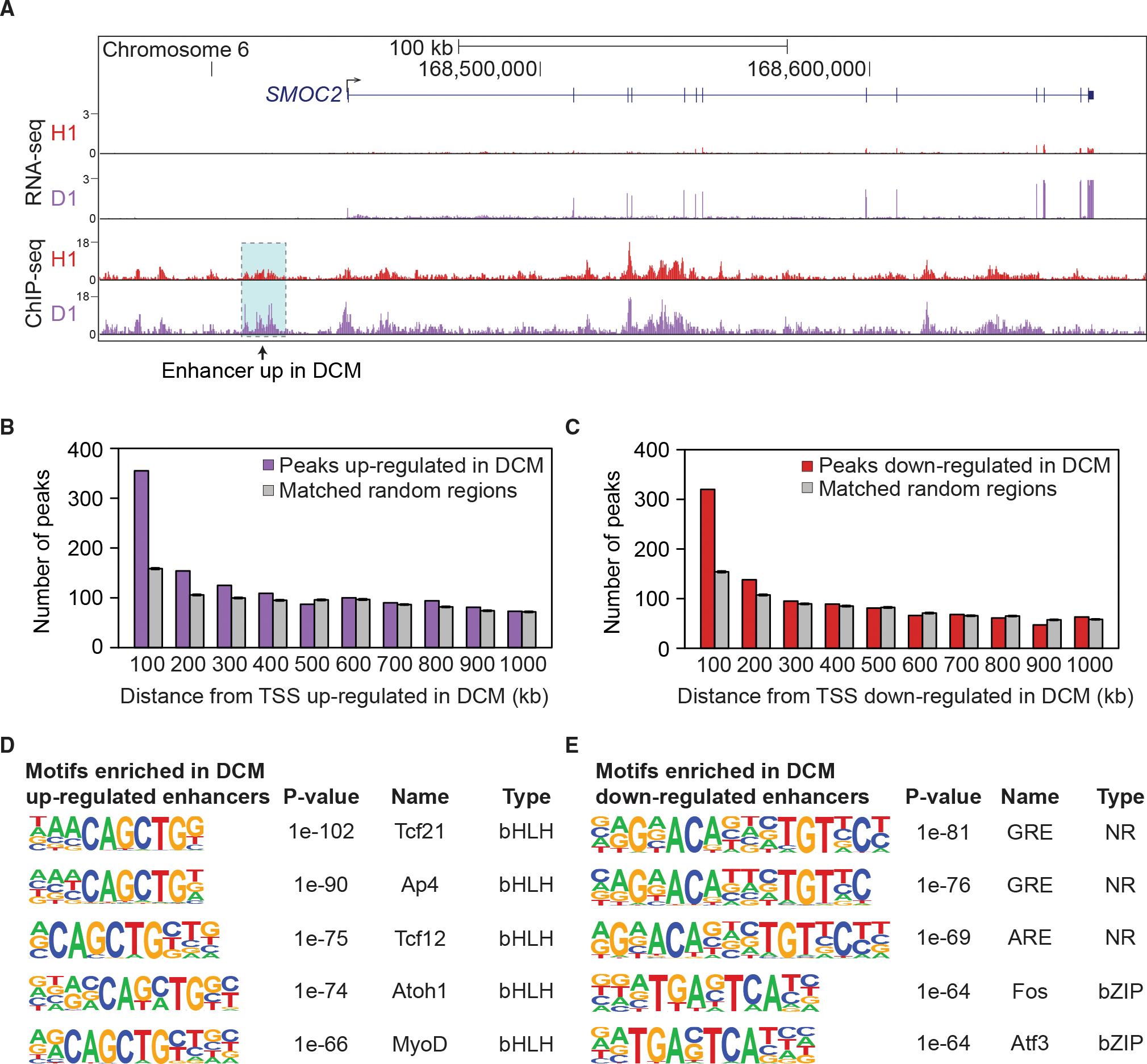
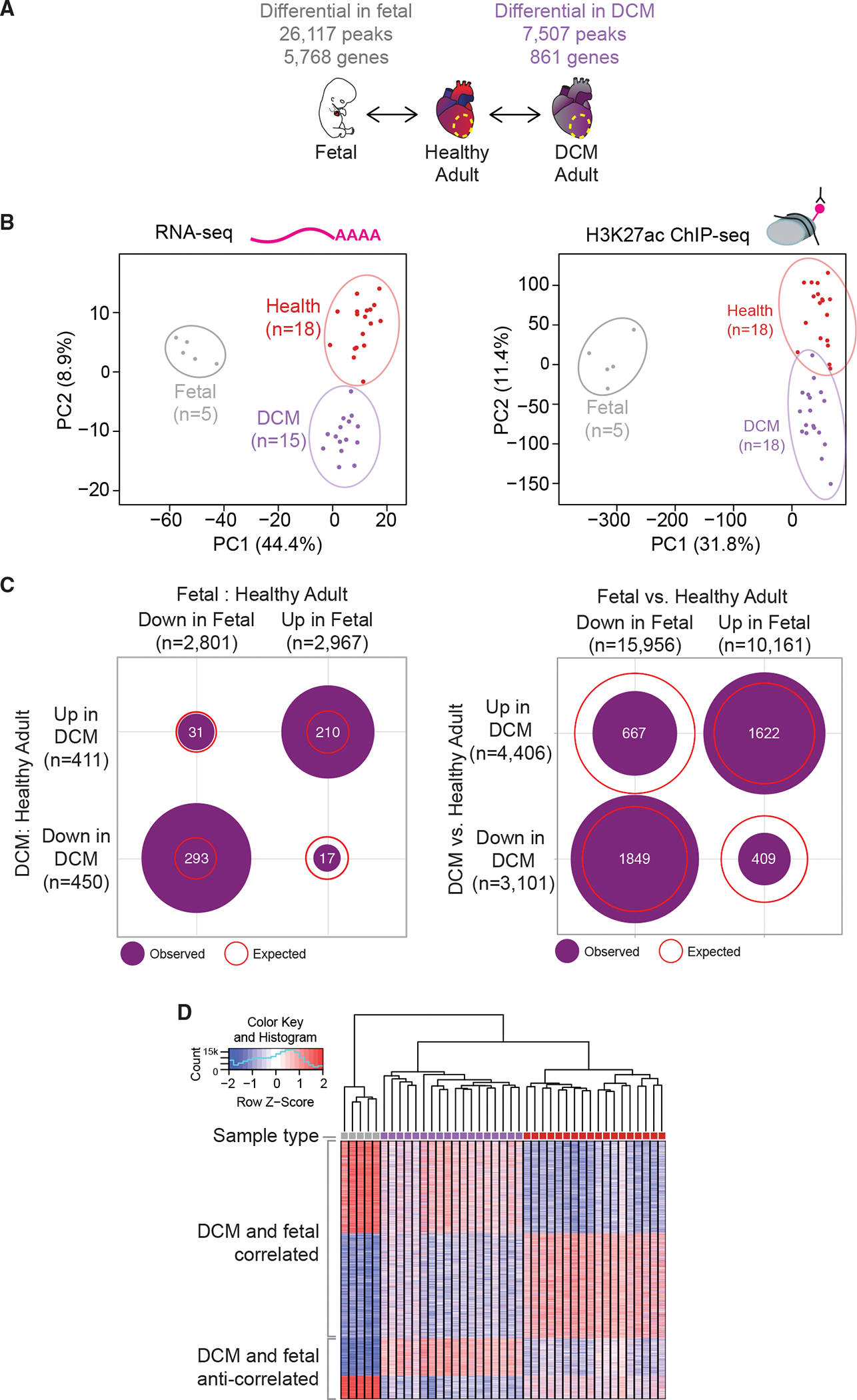
Heart disease is associated with re-expression of key transcription factors normally active only during prenatal development of the heart. However, the impact of this reactivation on the regulatory landscape in heart disease is unclear. Here, we use RNA-seq and ChIP-seq targeting a histone modification associated with active transcriptional enhancers to generate genome-wide enhancer maps from left ventricle tissue from up to 26 healthy controls, 18 individuals with idiopathic dilated cardiomyopathy (DCM), and five fetal hearts. Healthy individuals have a highly reproducible epigenomic landscape, consisting of more than 33,000 predicted heart enhancers. In contrast, we observe reproducible disease-associated changes in activity at 6,850 predicted heart enhancers. Combined analysis of adult and fetal samples reveals that the heart disease epigenome and transcriptome both acquire fetal-like characteristics, with 3,400 individual enhancers sharing fetal regulatory properties. We also provide a comprehensive data resource (http://heart.lbl.gov) for the mechanistic exploration of DCM etiology.
Keywords: CP: Molecular biology; RNA-seq; enhancers; fetalization; genomics; hIP-seq; heart disease; regulatory elements; transgenic assay.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Barth AS, Kuner R, Buness A, Ruschhaupt M, Merk S, Zwermann L, Kääb S, Kreuzer E, Steinbeck G, Mansmann U, et al. (2006). Identification of a common gene expression signature in dilated cardiomyopathy across independent microarray studies. J. Am. Coll. Cardiol. 48, 1610–1617. 10.1016/j.jacc.2006.07.026. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
