

CLN3 is required for the clearance of glycerophosphodiesters from lysosomes
- PMID: 36131016
- PMCID: PMC10510443
- DOI: 10.1038/s41586-022-05221-y
CLN3 is required for the clearance of glycerophosphodiesters from lysosomes
Abstract
Lysosomes have many roles, including degrading macromolecules and signalling to the nucleus1. Lysosomal dysfunction occurs in various human conditions, such as common neurodegenerative diseases and monogenic lysosomal storage disorders (LSDs)2-4. For most LSDs, the causal genes have been identified but, in some, the function of the implicated gene is unknown, in part because lysosomes occupy a small fraction of the cellular volume so that changes in lysosomal contents are difficult to detect. Here we develop the LysoTag mouse for the tissue-specific isolation of intact lysosomes that are compatible with the multimodal profiling of their contents. We used the LysoTag mouse to study CLN3, a lysosomal transmembrane protein with an unknown function. In children, the loss of CLN3 causes juvenile neuronal ceroid lipofuscinosis (Batten disease), a lethal neurodegenerative LSD. Untargeted metabolite profiling of lysosomes from the brains of mice lacking CLN3 revealed a massive accumulation of glycerophosphodiesters (GPDs)-the end products of glycerophospholipid catabolism. GPDs also accumulate in the lysosomes of CLN3-deficient cultured cells and we show that CLN3 is required for their lysosomal egress. Loss of CLN3 also disrupts glycerophospholipid catabolism in the lysosome. Finally, we found elevated levels of glycerophosphoinositol in the cerebrospinal fluid of patients with Batten disease, suggesting the potential use of glycerophosphoinositol as a disease biomarker. Our results show that CLN3 is required for the lysosomal clearance of GPDs and reveal Batten disease as a neurodegenerative LSD with a defect in glycerophospholipid metabolism.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing Interests
M.A-R. is a scientific advisory board member of Lycia Therapeutics. A.D.D. and F.D.P. have a collaborative research agreement with Amicus Therapeutics, Inc. All other authors declare no competing interests.
Figures
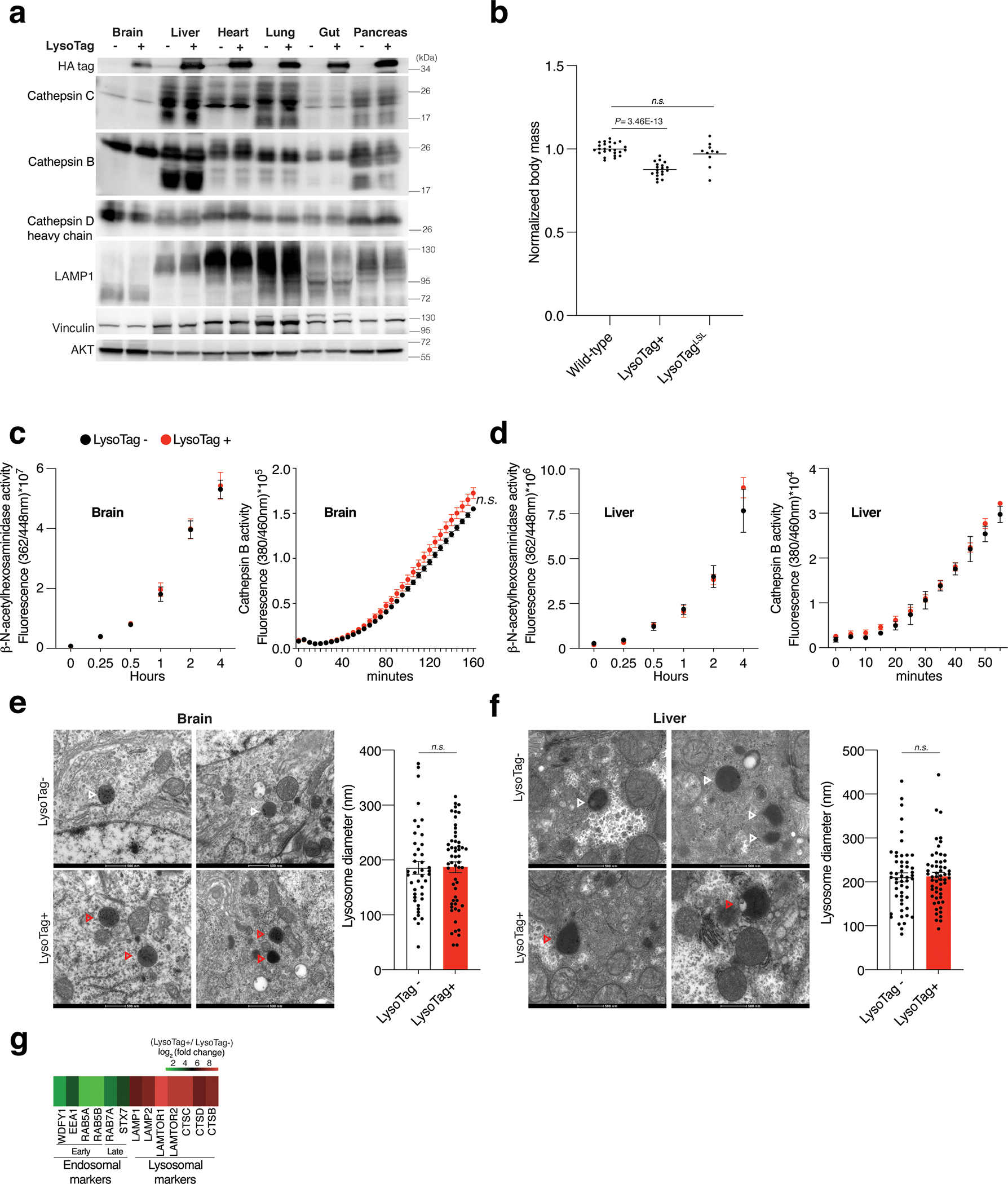
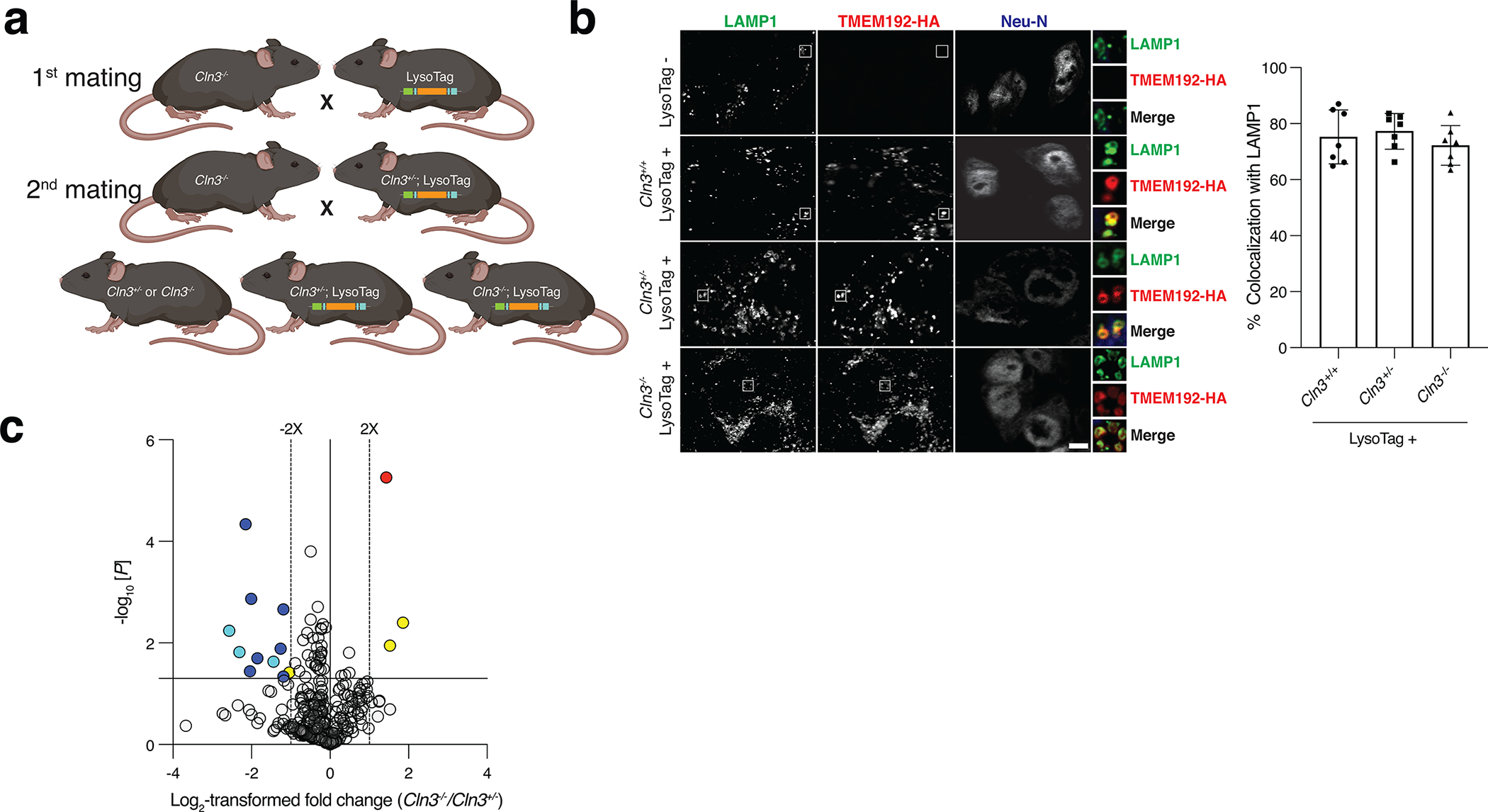
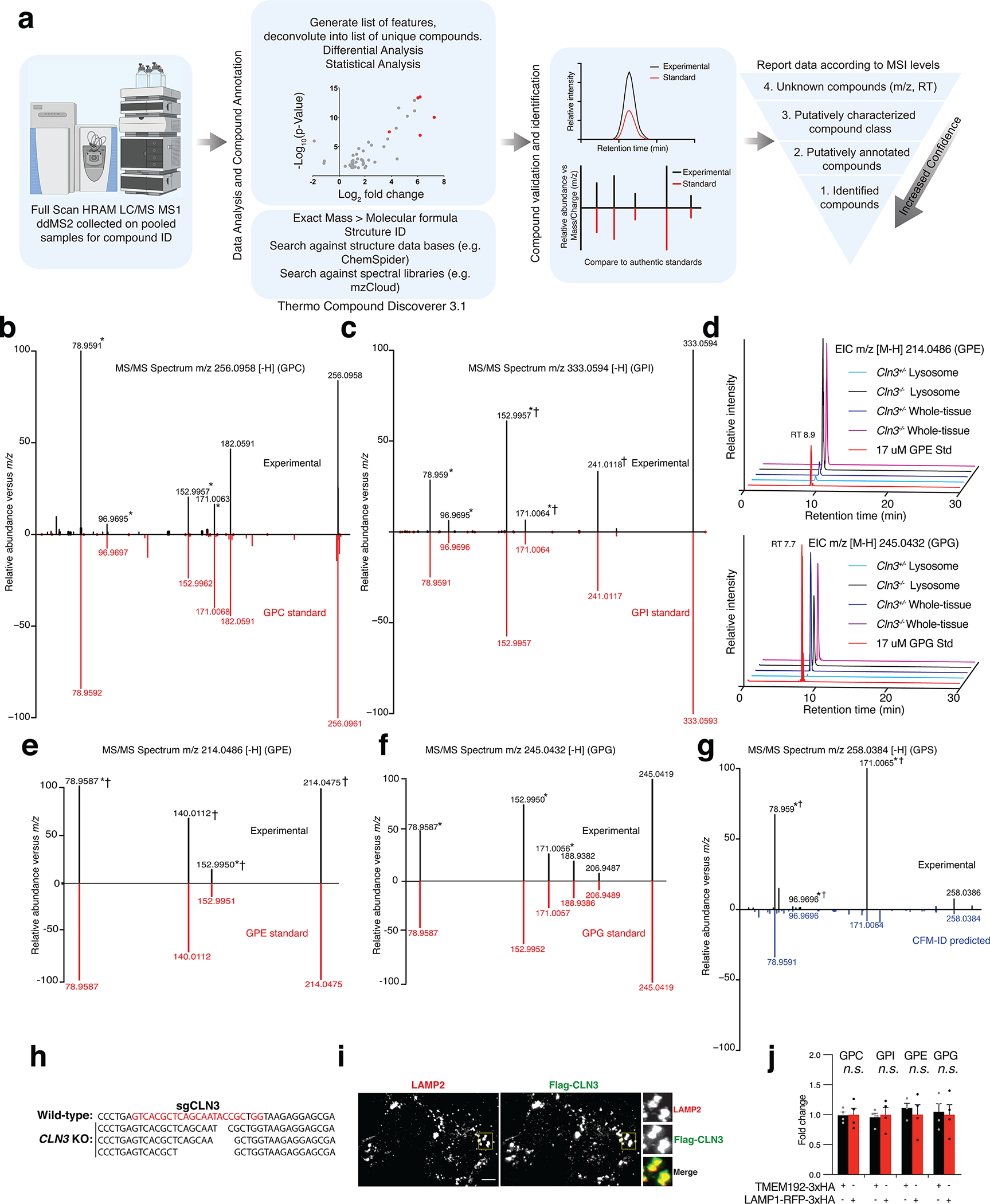
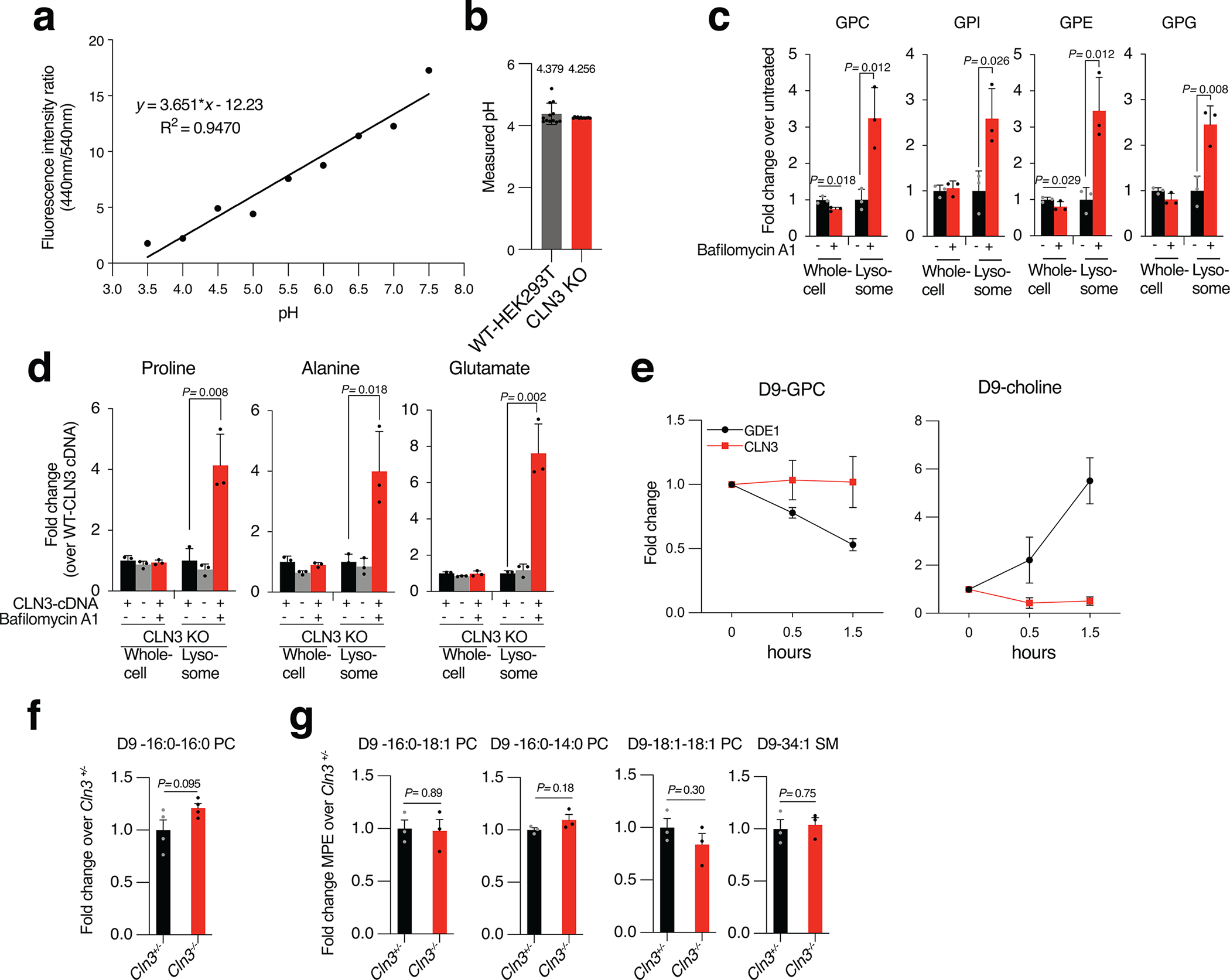
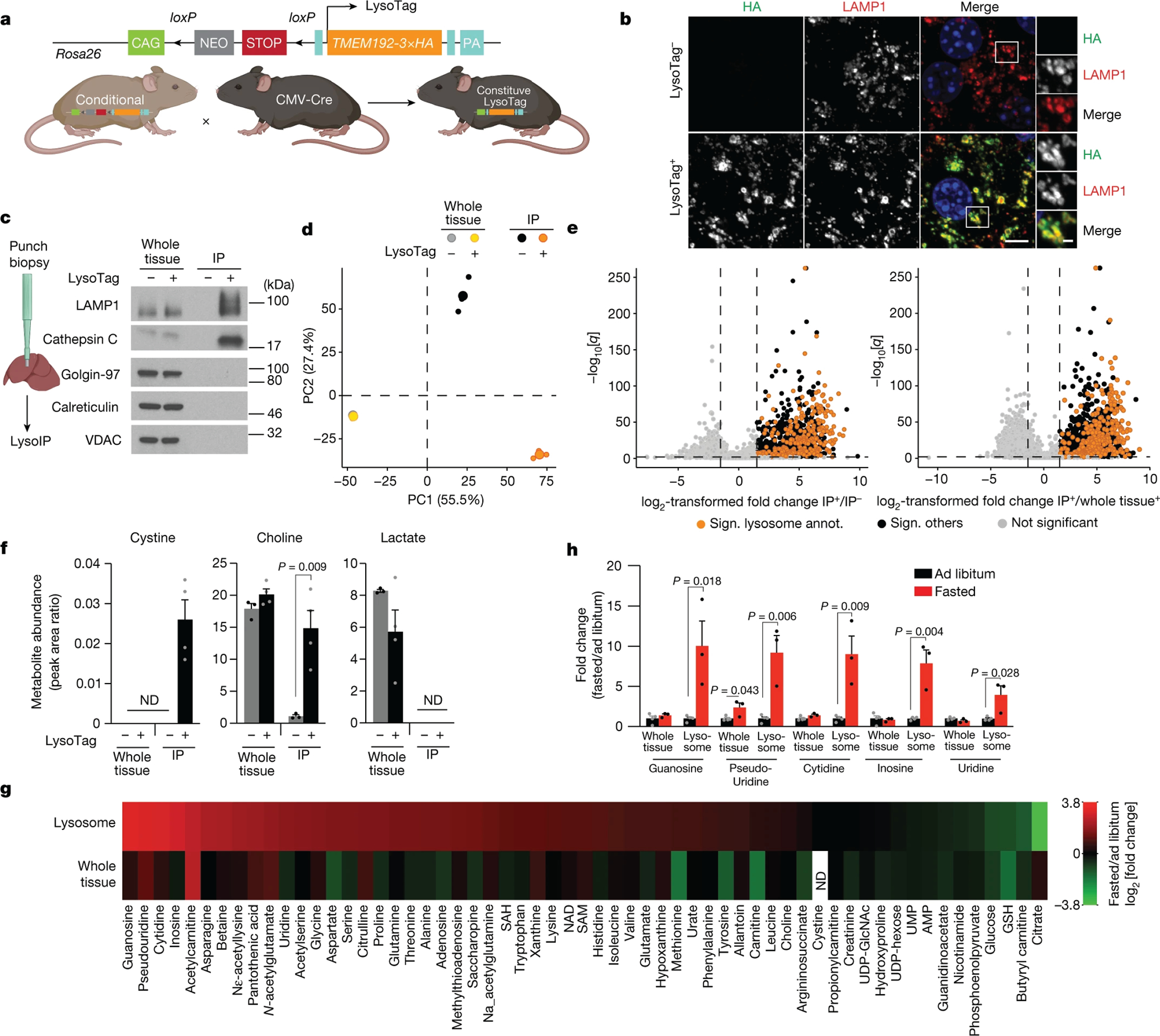
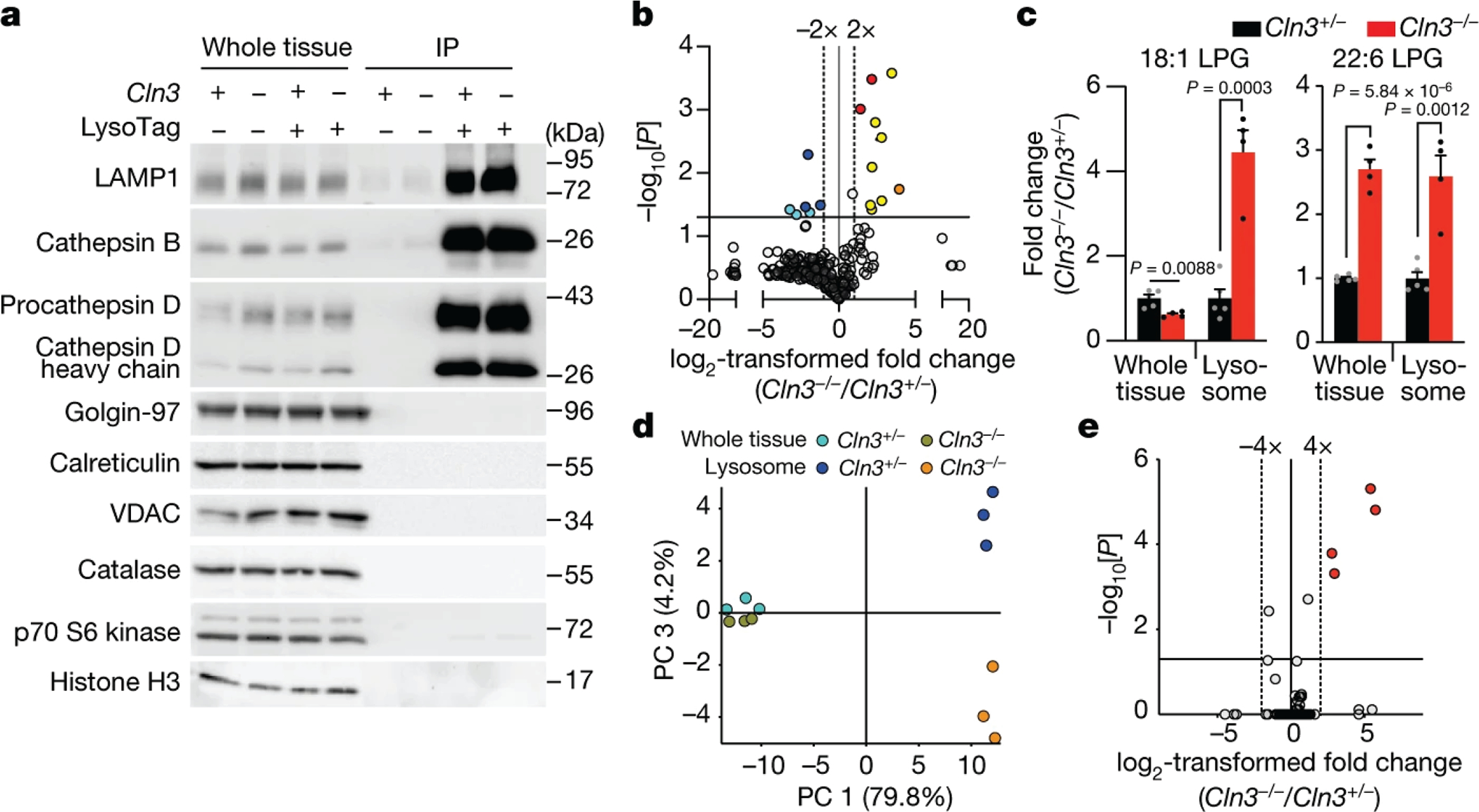
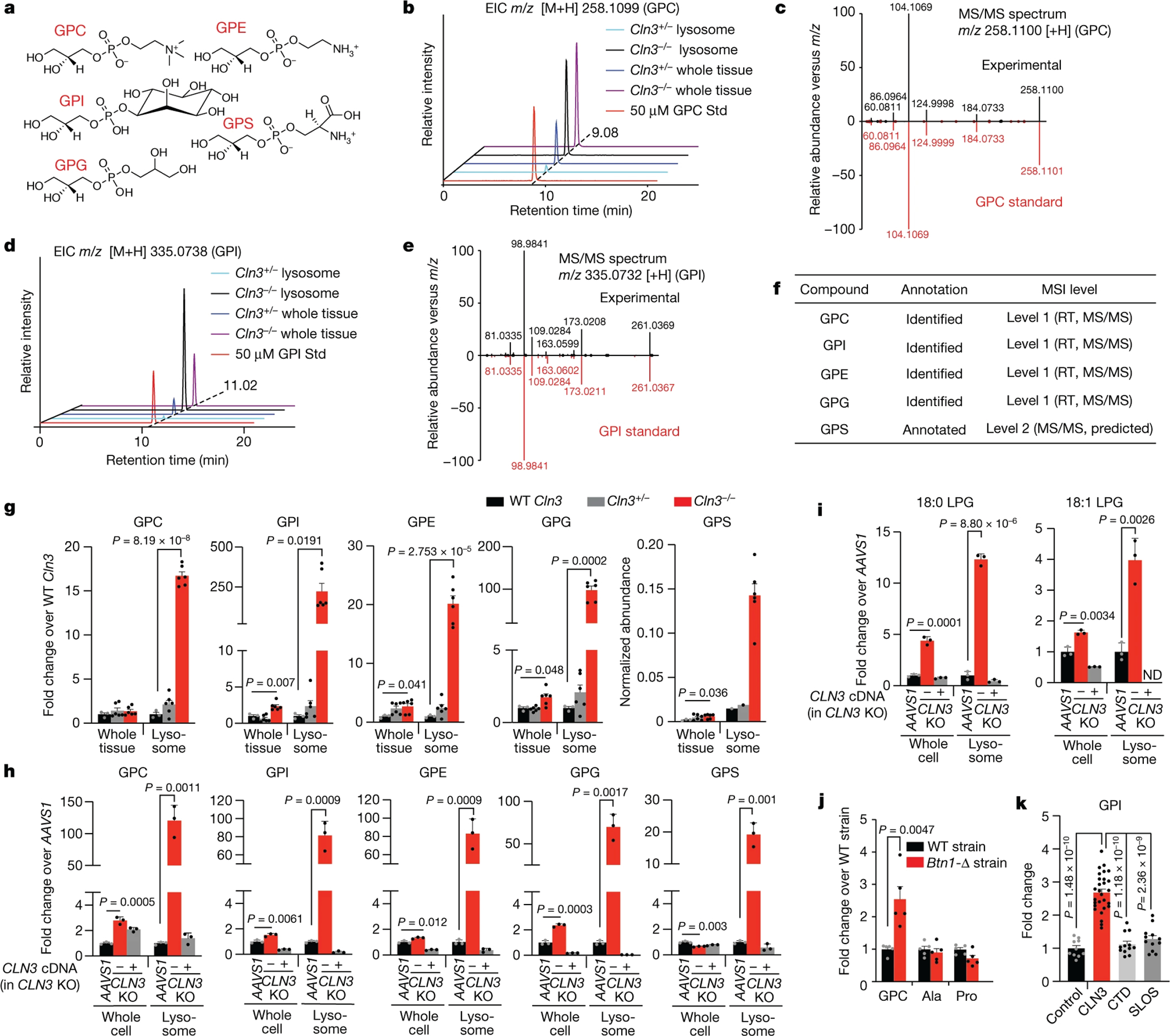
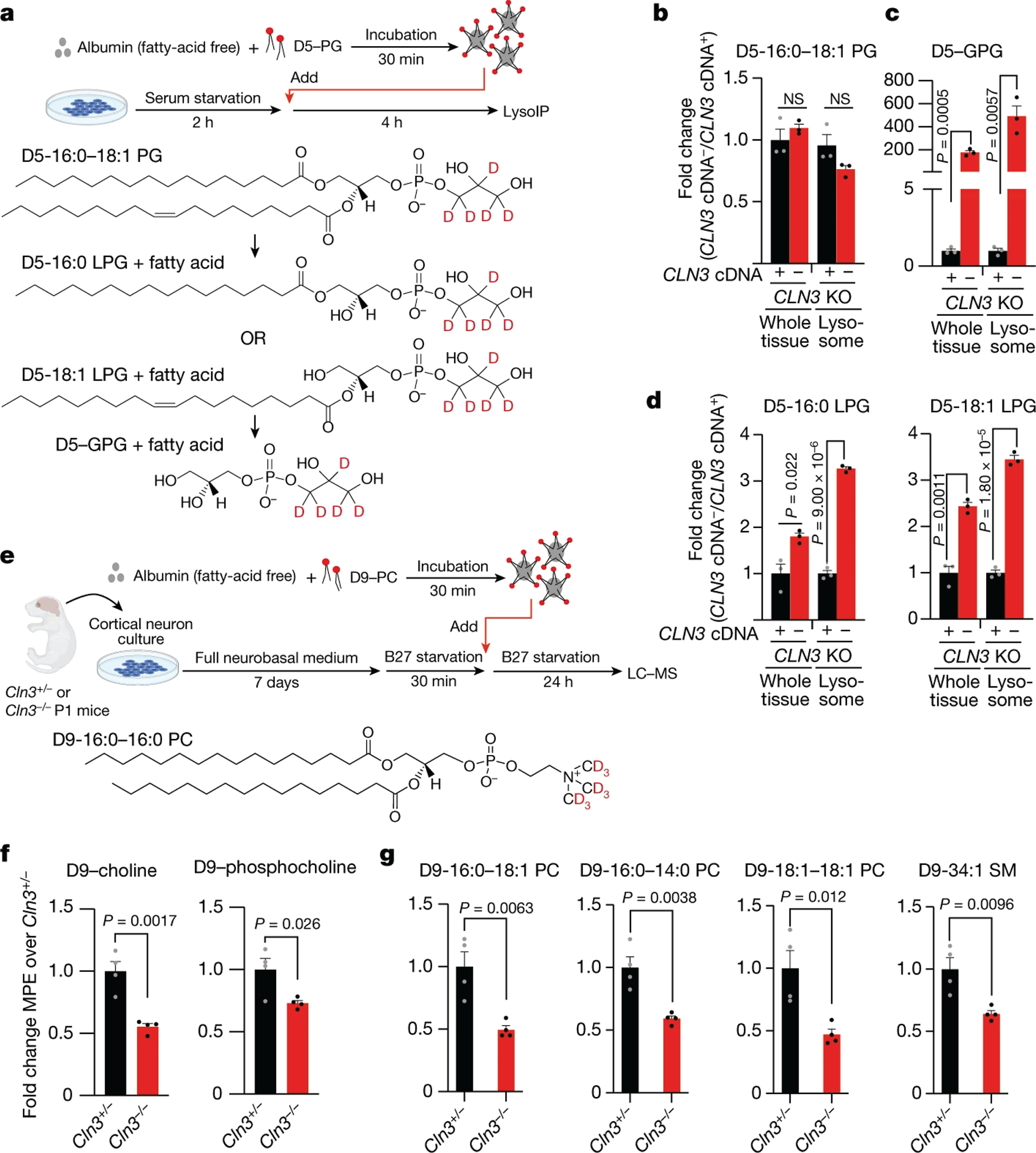
References
-
- Medoh UN, Chen JY & Abu-Remaileh M Lessons from metabolic perturbations in lysosomal storage disorders for neurodegeneration. Current Opinion in Systems Biology 29, 100408, doi:10.1016/j.coisb.2021.100408 (2022). - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
