

A cellular hierarchy in melanoma uncouples growth and metastasis
- PMID: 36131018
- PMCID: PMC10439739
- DOI: 10.1038/s41586-022-05242-7
A cellular hierarchy in melanoma uncouples growth and metastasis
Erratum in
-
Author Correction: A cellular hierarchy in melanoma uncouples growth and metastasis.Nature. 2022 Nov;611(7934):E4. doi: 10.1038/s41586-022-05415-4. Nature. 2022. PMID: 36261534 No abstract available.
Abstract
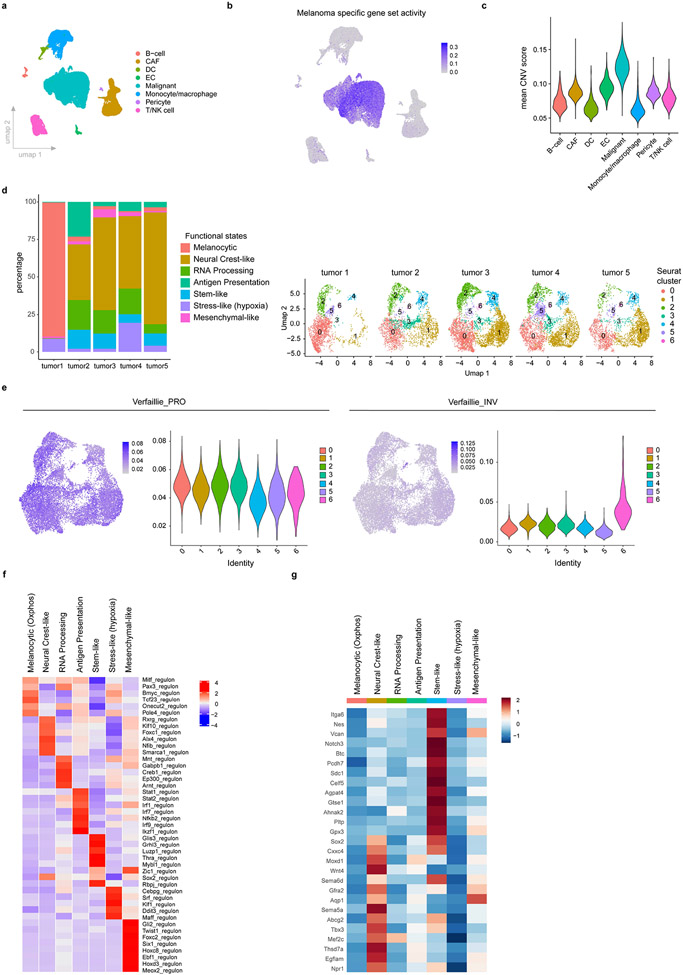
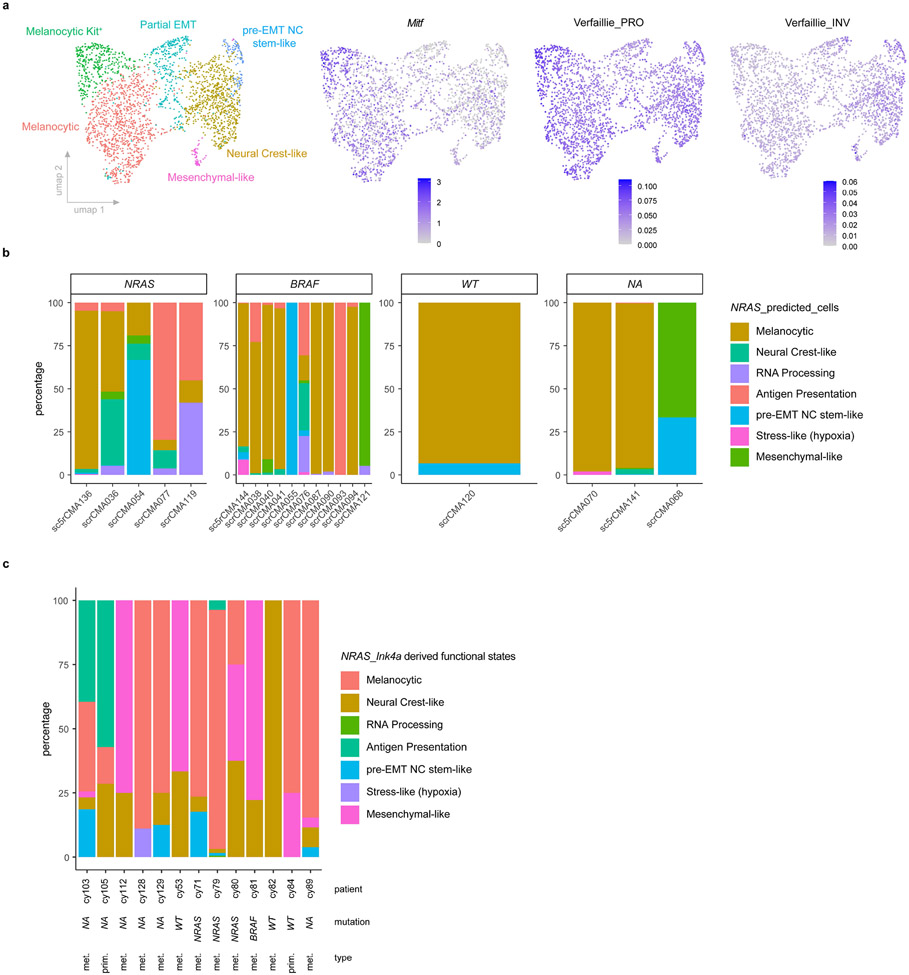
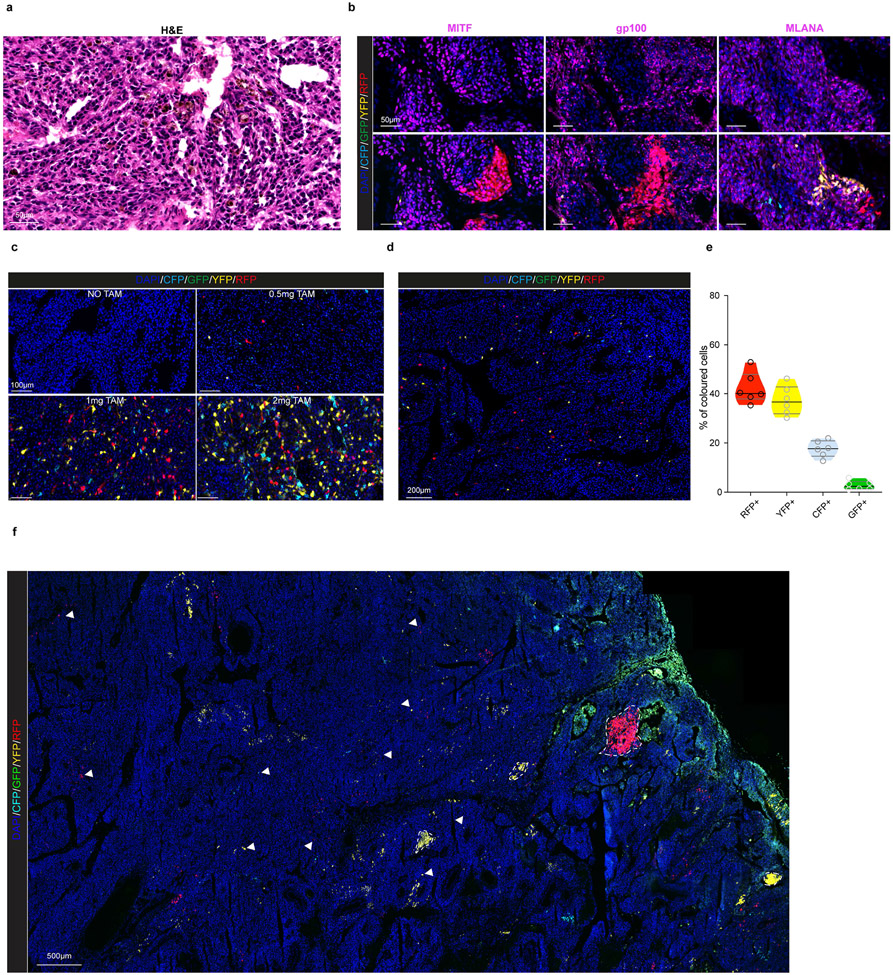
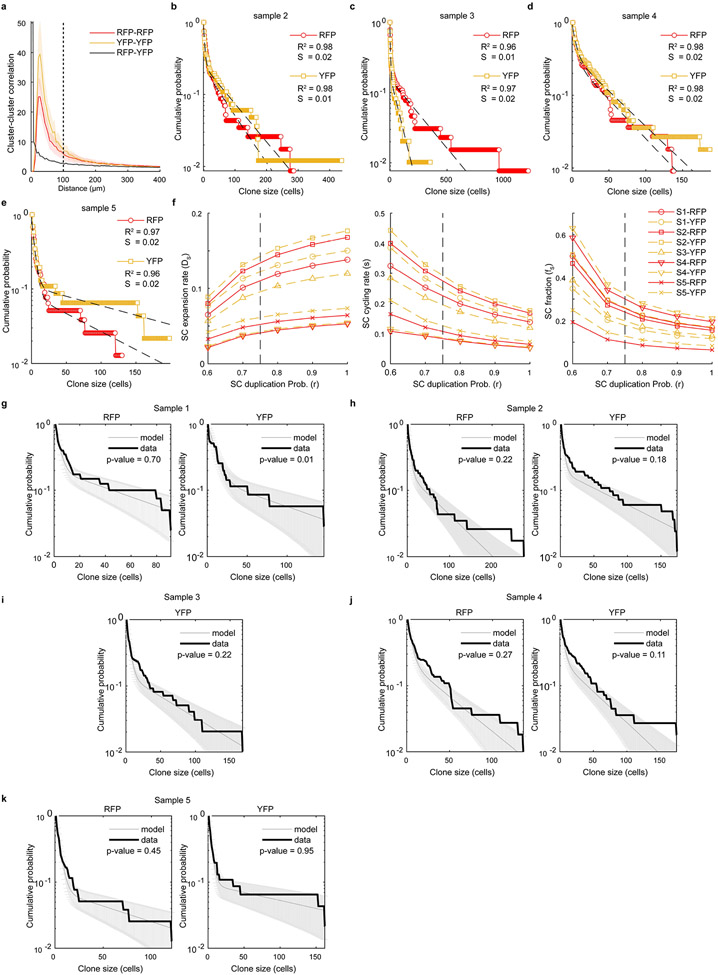
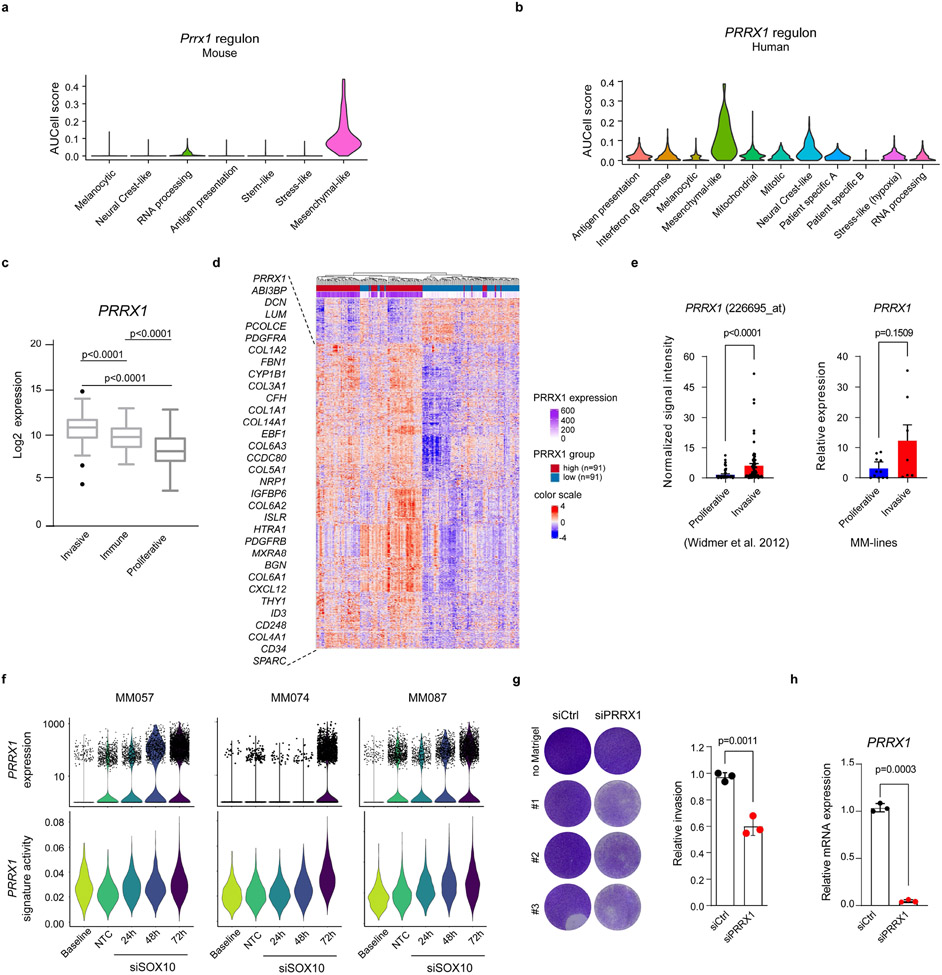
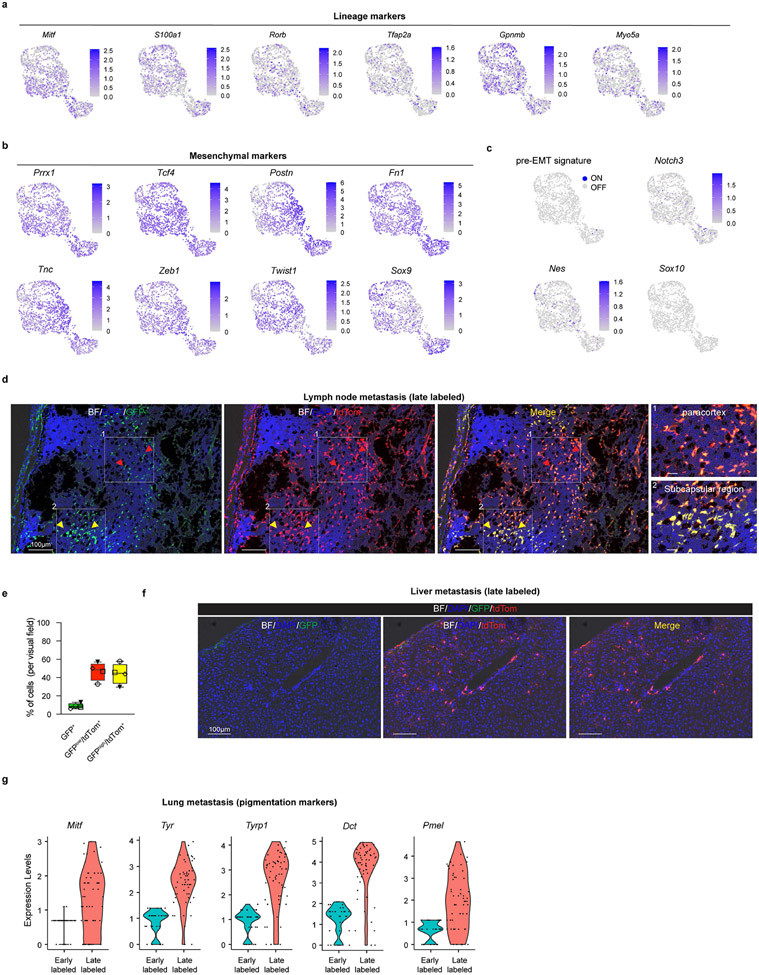
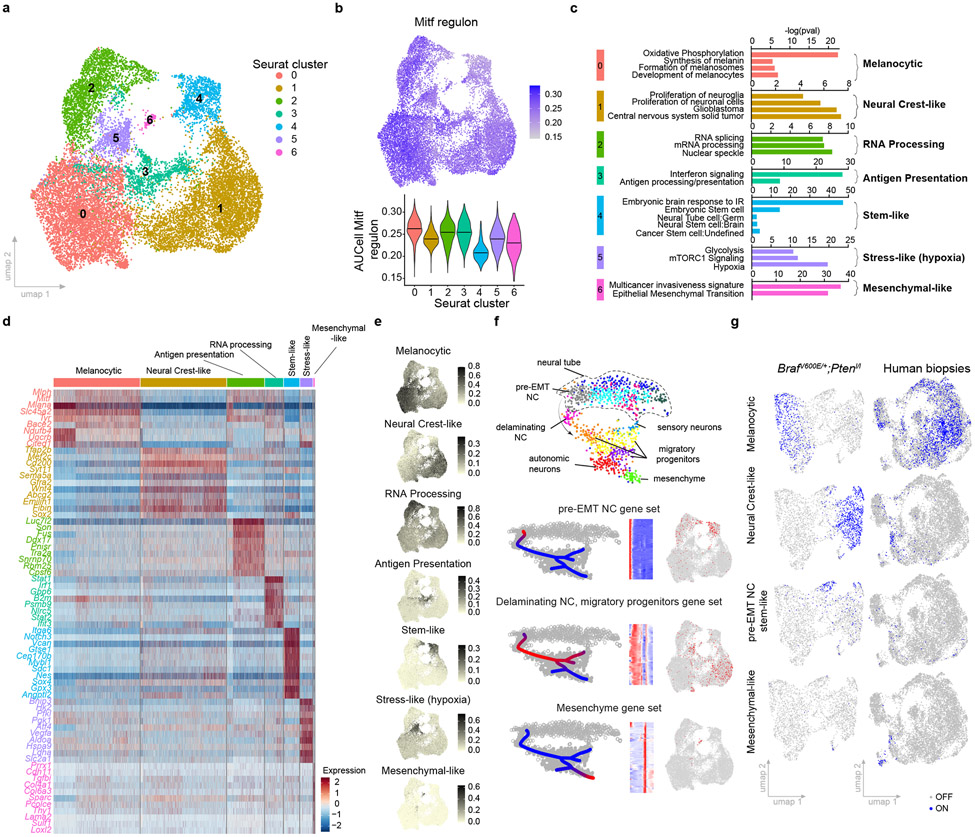
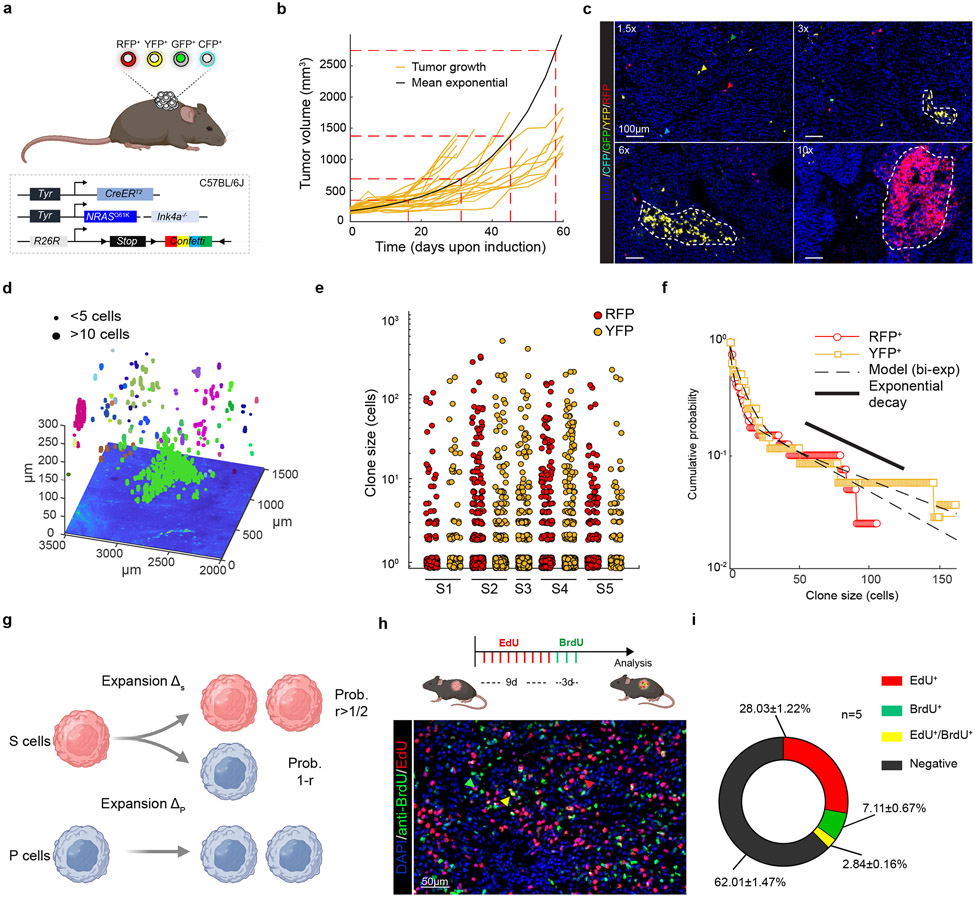
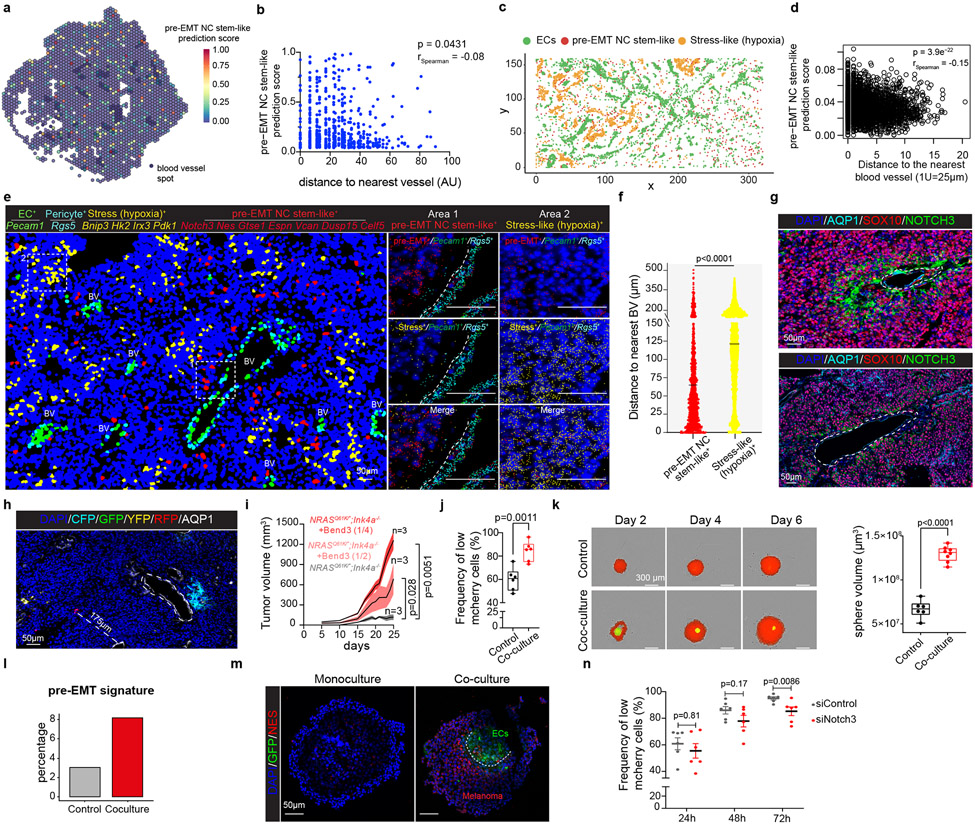
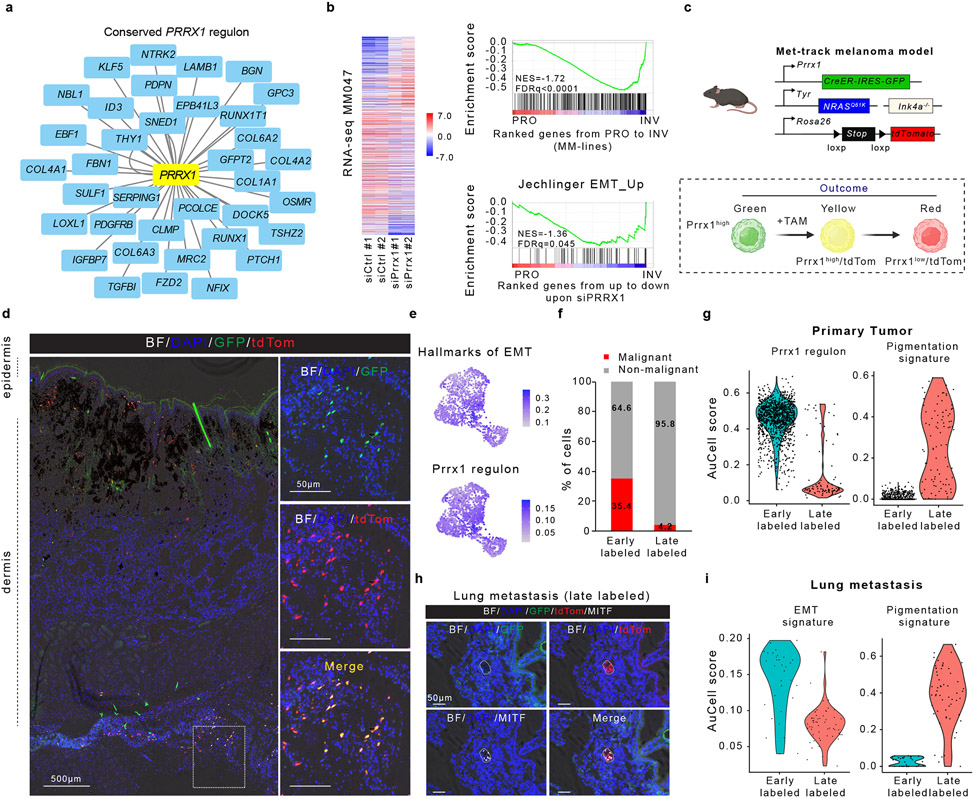
Although melanoma is notorious for its high degree of heterogeneity and plasticity1,2, the origin and magnitude of cell-state diversity remains poorly understood. Equally, it is unclear whether growth and metastatic dissemination are supported by overlapping or distinct melanoma subpopulations. Here, by combining mouse genetics, single-cell and spatial transcriptomics, lineage tracing and quantitative modelling, we provide evidence of a hierarchical model of tumour growth that mirrors the cellular and molecular logic underlying the cell-fate specification and differentiation of the embryonic neural crest. We show that tumorigenic competence is associated with a spatially localized perivascular niche, a phenotype acquired through an intercellular communication pathway established by endothelial cells. Consistent with a model in which only a fraction of cells are fated to fuel growth, temporal single-cell tracing of a population of melanoma cells with a mesenchymal-like state revealed that these cells do not contribute to primary tumour growth but, instead, constitute a pool of metastatic initiating cells that switch cell identity while disseminating to secondary organs. Our data provide a spatially and temporally resolved map of the diversity and trajectories of melanoma cell states and suggest that the ability to support growth and metastasis are limited to distinct pools of cells. The observation that these phenotypic competencies can be dynamically acquired after exposure to specific niche signals warrant the development of therapeutic strategies that interfere with the cancer cell reprogramming activity of such microenvironmental cues.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Figures
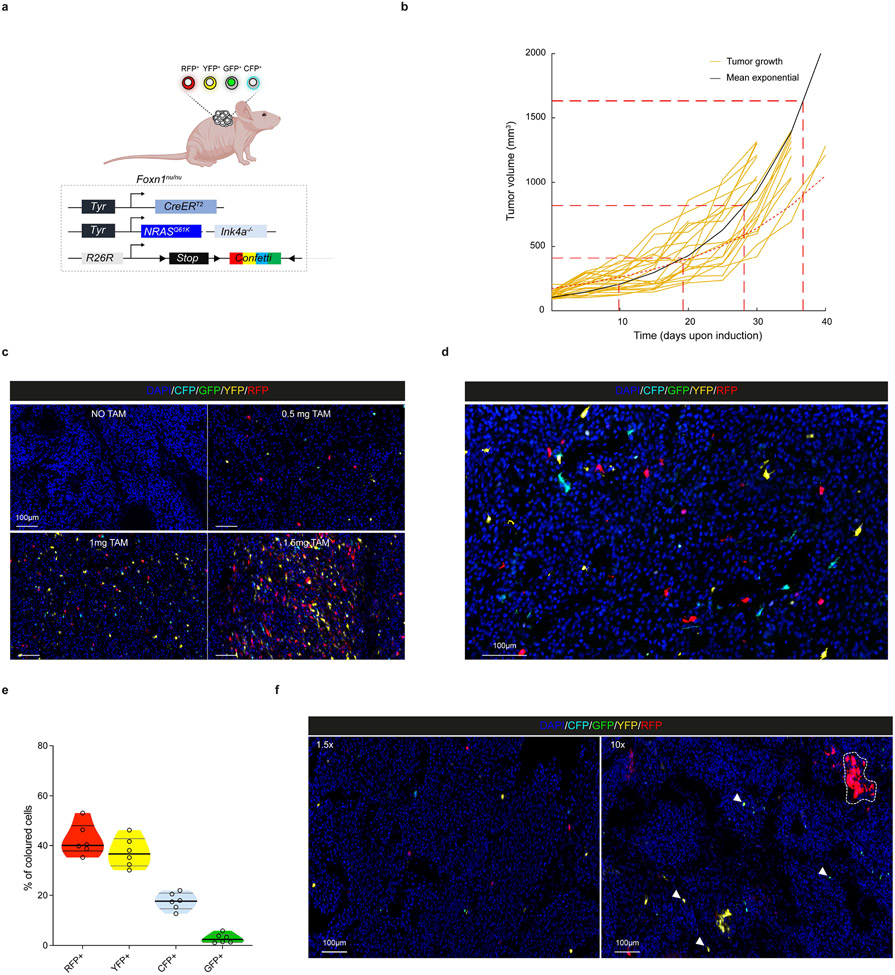
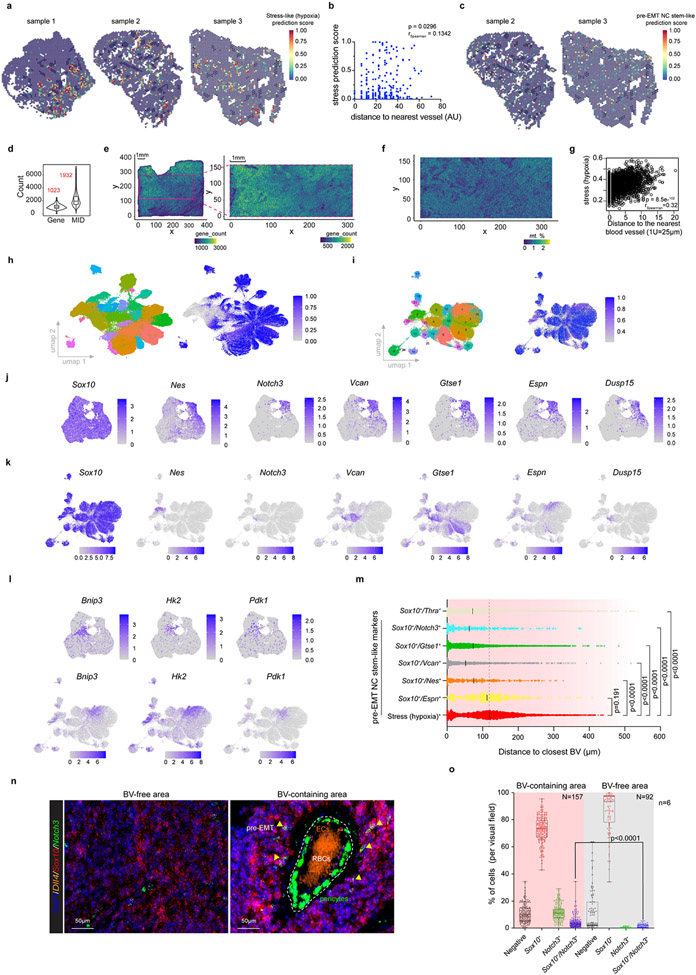
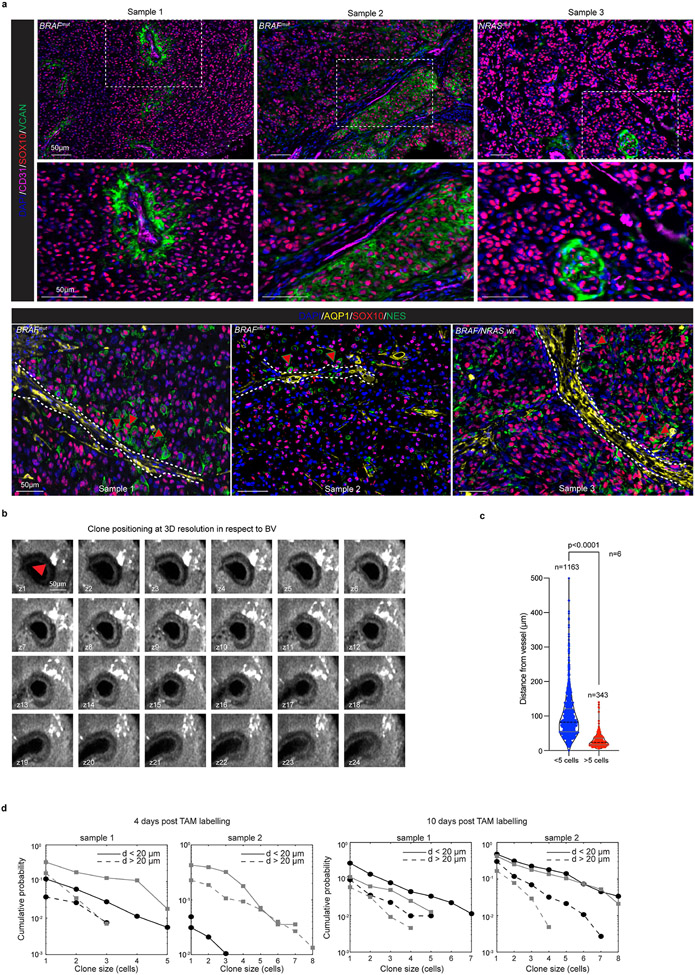
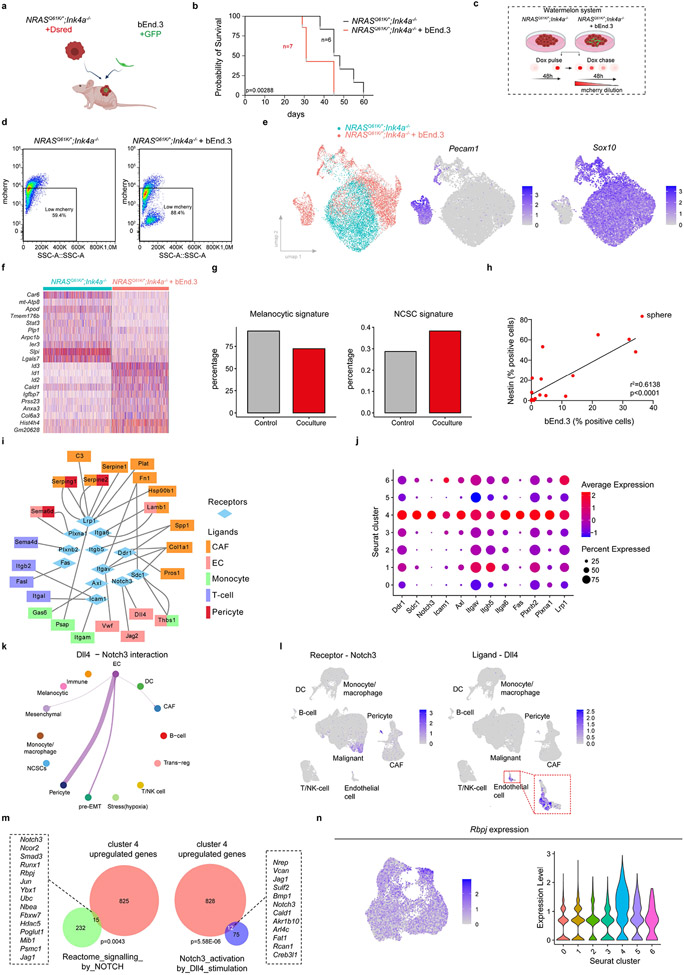
Comment in
-
The hierarchy of melanoma.Nat Rev Cancer. 2022 Dec;22(12):658. doi: 10.1038/s41568-022-00532-8. Nat Rev Cancer. 2022. PMID: 36319698 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
