

Resistance to tyrosine kinase inhibitors promotes renal cancer progression through MCPIP1 tumor-suppressor downregulation and c-Met activation
- PMID: 36138026
- PMCID: PMC9500022
- DOI: 10.1038/s41419-022-05251-4
Resistance to tyrosine kinase inhibitors promotes renal cancer progression through MCPIP1 tumor-suppressor downregulation and c-Met activation
Abstract
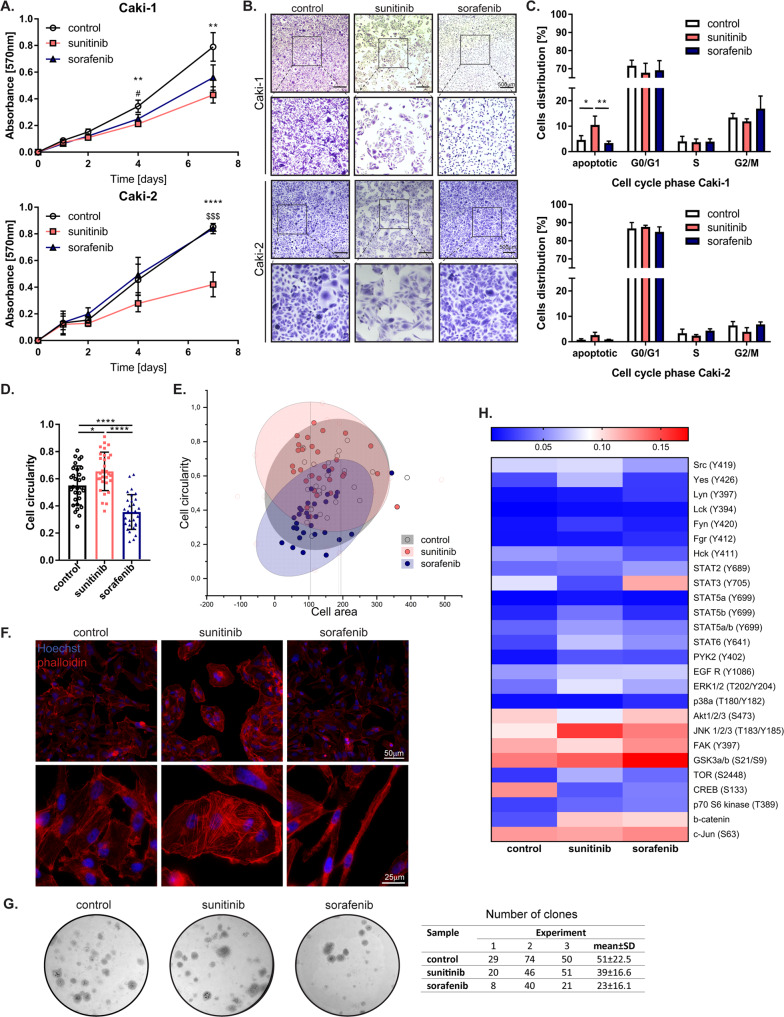
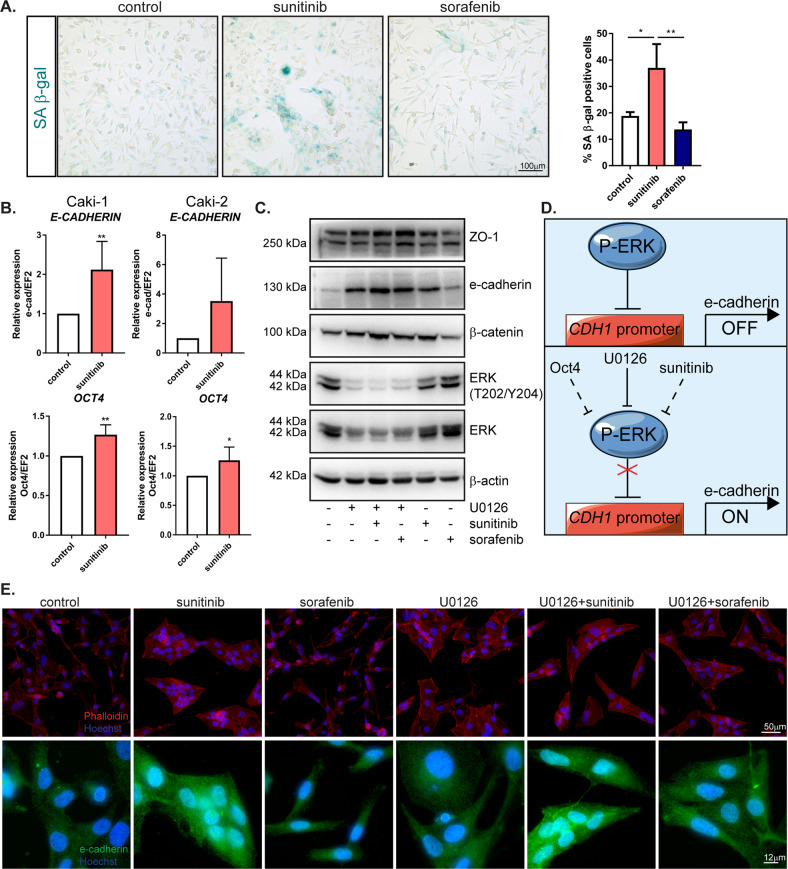
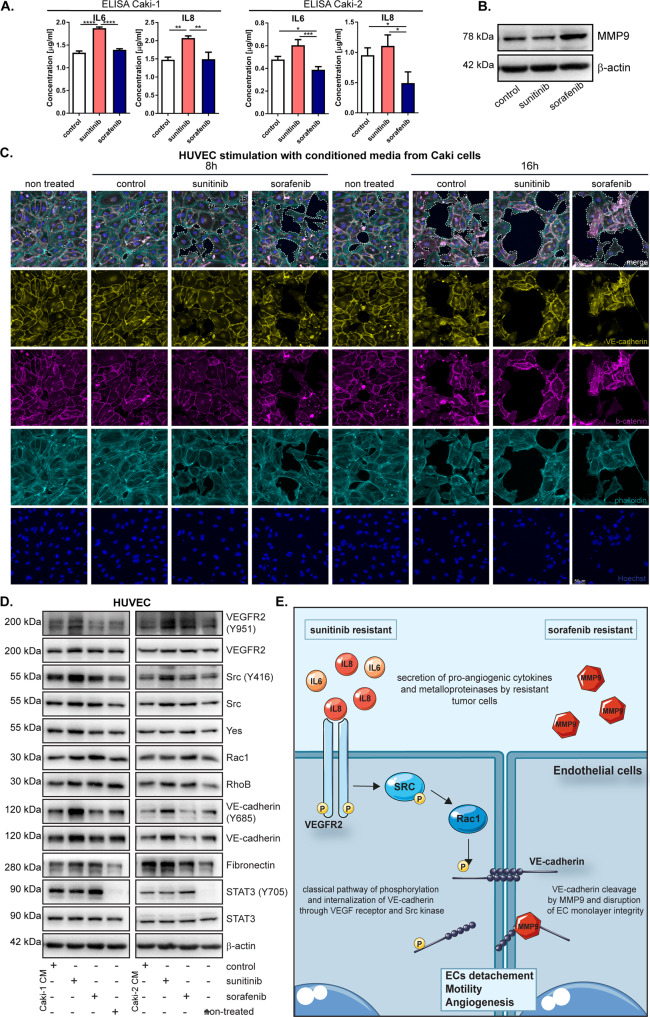
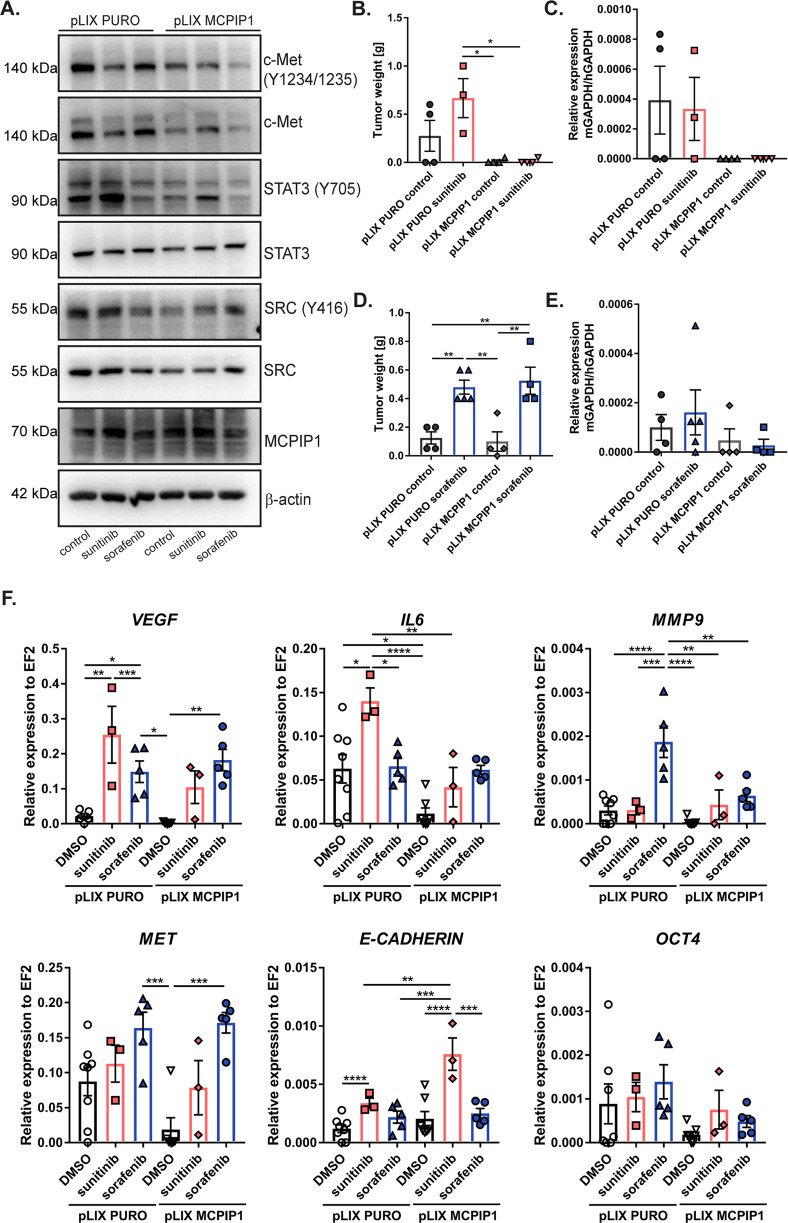
Tyrosine kinase inhibitors (TKIs) are the most commonly used targeted therapeutics in clear-cell renal cell carcinoma (ccRCC); however, drug resistance limits their utility and can lead to tumor "flare-up" and progression. In this study, we show that RCC resistance to sunitinib and sorafenib involves different mechanisms and leads to increased malignancy. Sunitinib decreased tumor growth and cell motility along with increased E-cadherin expression and secretion of the proangiogenic cytokines IL6 and IL8, which activated senescence in ccRCC cells and led to VE-cadherin phosphorylation, enhancing tumor angiogenesis. Sorafenib resistance increased the levels of mesenchymal markers and the secretion of MMP9, which cleaved VE-cadherin and disrupted endothelial cell integrity. Both sunitinib resistance and sorafenib resistance led to activation of the c-Met receptor IRAK1 and downregulation of the tumor suppressor MCPIP1, resulting in an increase in the metastasis of resistant cells, possibly due in part to enhanced vascularization of ccRCC. MCPIP1 overexpression partially overcame resistance to these drugs by decreasing micrometastasis and decreasing the expression of factors involved in tumorigenesis. In tumor samples from ccRCC patients, we observed a significant increase in the level of the c-Met receptor, IRAK1 and a decrease in MCPIP1 with respect to normal kidney tissue. Our results indicate separate novel mechanisms for sunitinib and sorafenib resistance, which both lead to MCPIP1 inhibition and ccRCC progression. The presented study suggests caution in the treatment of RCC with TKIs, which may lead to the unintended outcome of tumor progression.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Motzer RJ, Escudier B, Tomczak P, Hutson TE, Michaelson MD, Negrier S, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013;14:552–62. doi: 10.1016/S1470-2045(13)70093-7. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
