

Identification and in-silico characterization of splice-site variants from a large cardiogenetic national registry
- PMID: 36138163
- PMCID: PMC10172209
- DOI: 10.1038/s41431-022-01193-9
Identification and in-silico characterization of splice-site variants from a large cardiogenetic national registry
Abstract
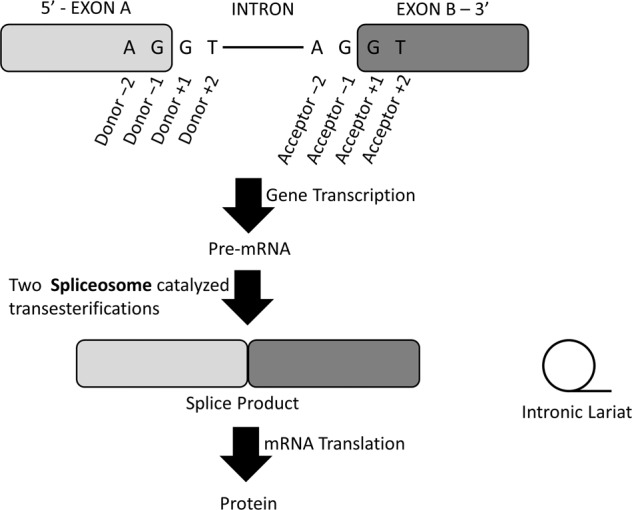
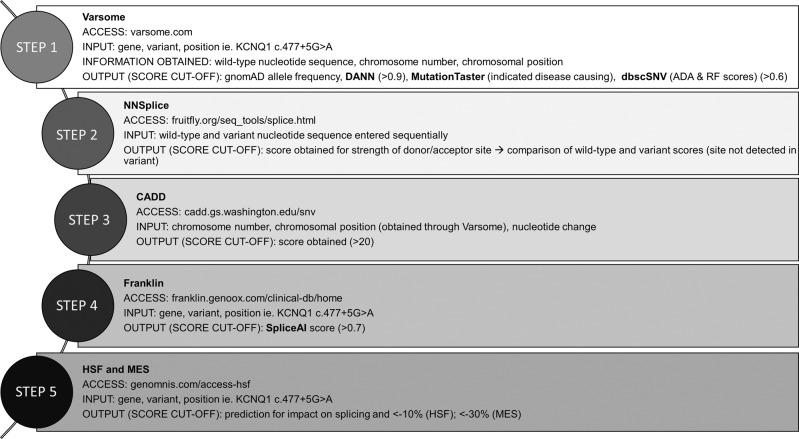
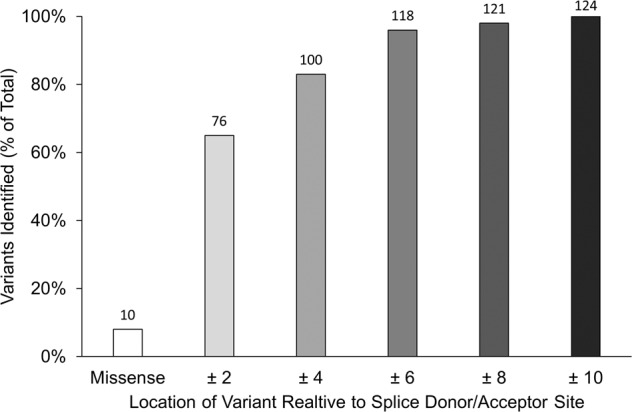
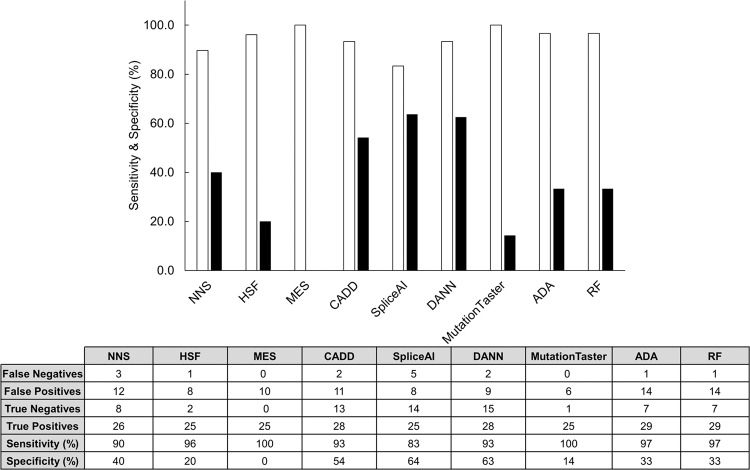
Splice-site variants in cardiac genes may predispose carriers to potentially lethal arrhythmias. To investigate, we screened 1315 probands and first-degree relatives enrolled in the Canadian Hearts in Rhythm Organization (HiRO) registry. 10% (134/1315) of patients in the HiRO registry carry variants within 10 base-pairs of the intron-exon boundary with 78% (104/134) otherwise genotype negative. These 134 probands were carriers of 57 unique variants. For each variant, American College of Medical Genetics and Genomics (ACMG) classification was revisited based on consensus between nine in silico tools. Due in part to the in silico algorithms, seven variants were reclassified from the original report, with the majority (6/7) downgraded. Our analyses predicted 53% (30/57) of variants to be likely/pathogenic. For the 57 variants, an average of 9 tools were able to score variants within splice sites, while 6.5 tools responded for variants outside these sites. With likely/pathogenic classification considered a positive outcome, the ACMG classification was used to calculate sensitivity/specificity of each tool. Among these, Combined Annotation Dependent Depletion (CADD) had good sensitivity (93%) and the highest response rate (131/134, 98%), dbscSNV was also sensitive (97%), and SpliceAI was the most specific (64%) tool. Splice variants remain an important consideration in gene elusive inherited arrhythmia syndromes. Screening for intronic variants, even when restricted to the ±10 positions as performed here may improve genetic testing yield. We compare 9 freely available in silico tools and provide recommendations regarding their predictive capabilities. Moreover, we highlight several novel cardiomyopathy-associated variants which merit further study.
© 2022. The Author(s), under exclusive licence to European Society of Human Genetics.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
