

Modular cytokine receptor-targeting chimeras for targeted degradation of cell surface and extracellular proteins
- PMID: 36138170
- PMCID: PMC9931583
- DOI: 10.1038/s41587-022-01456-2
Modular cytokine receptor-targeting chimeras for targeted degradation of cell surface and extracellular proteins
Abstract
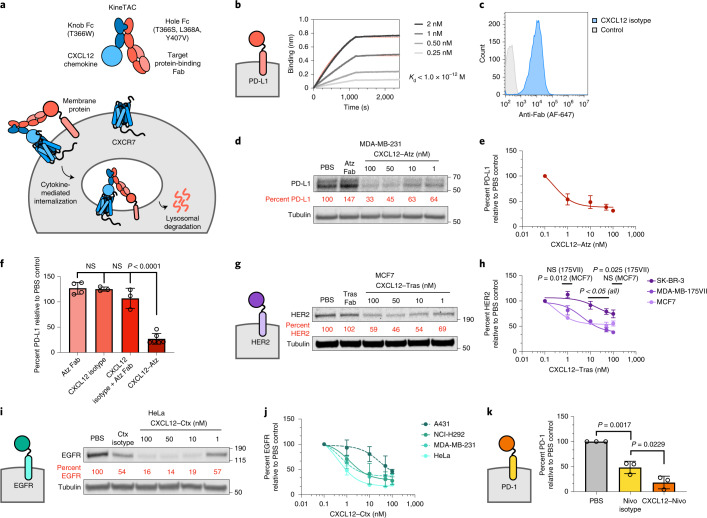
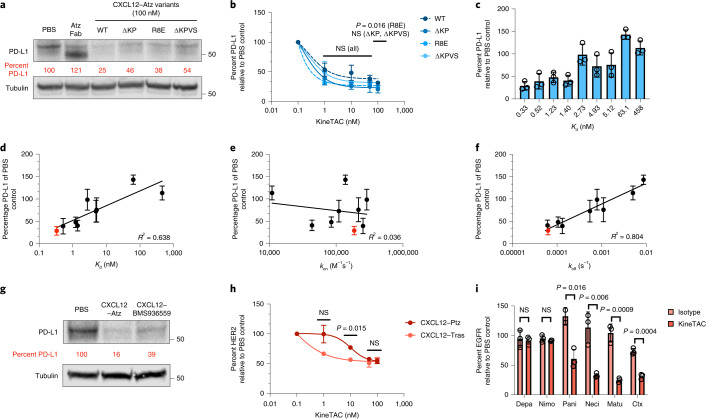
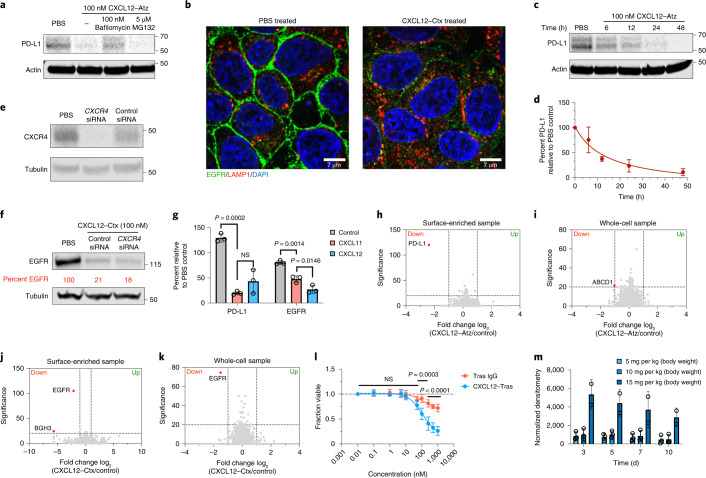
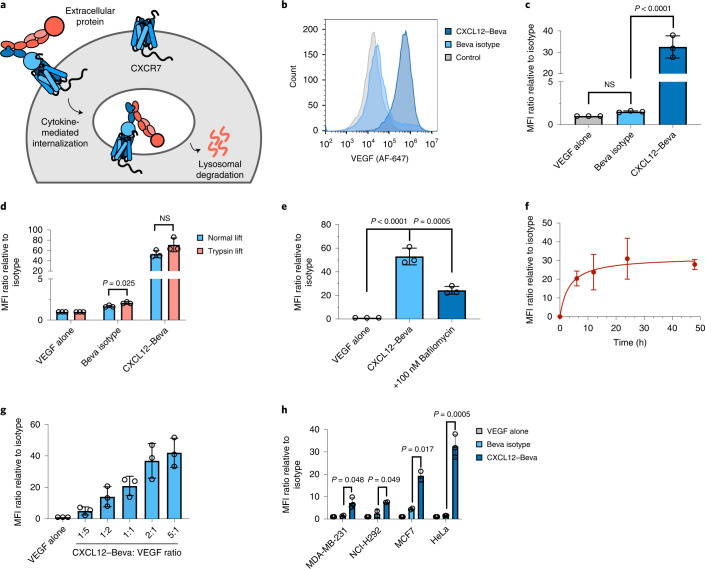
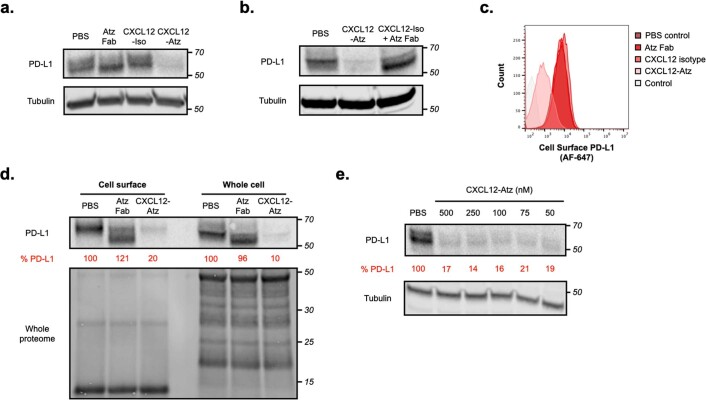
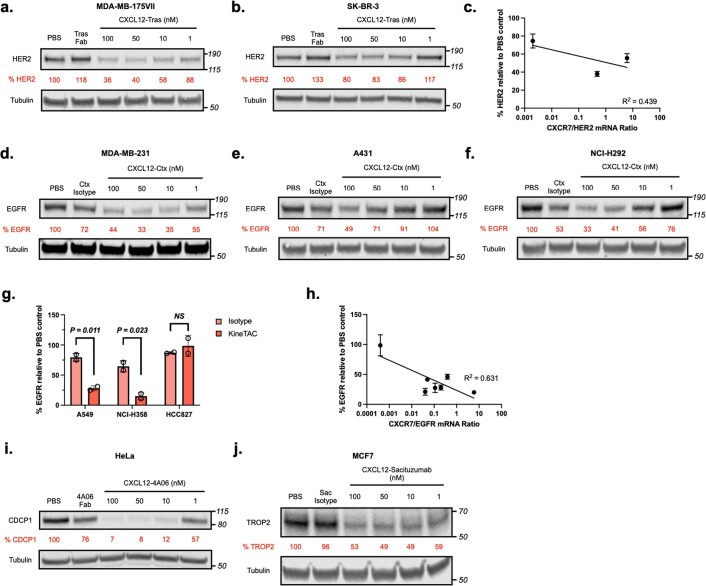
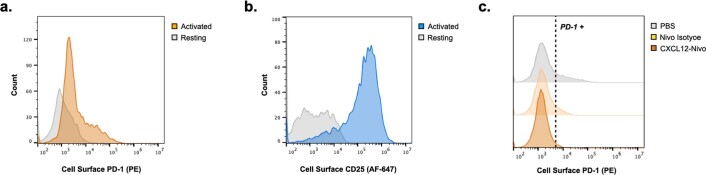
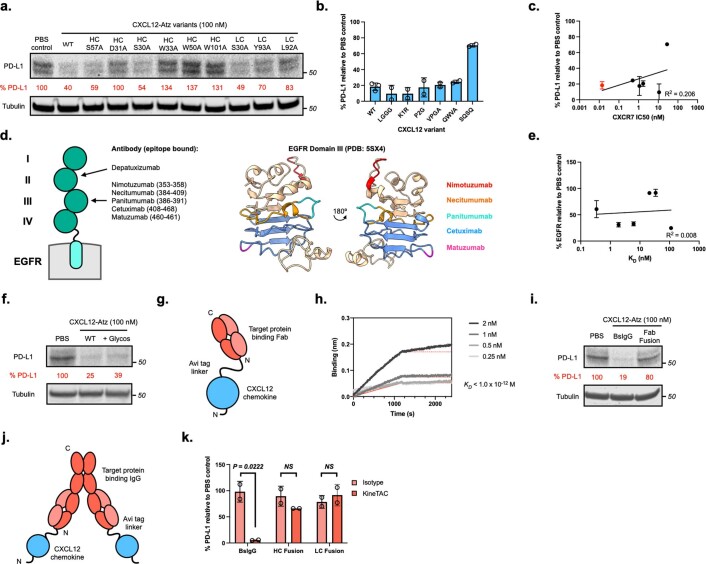
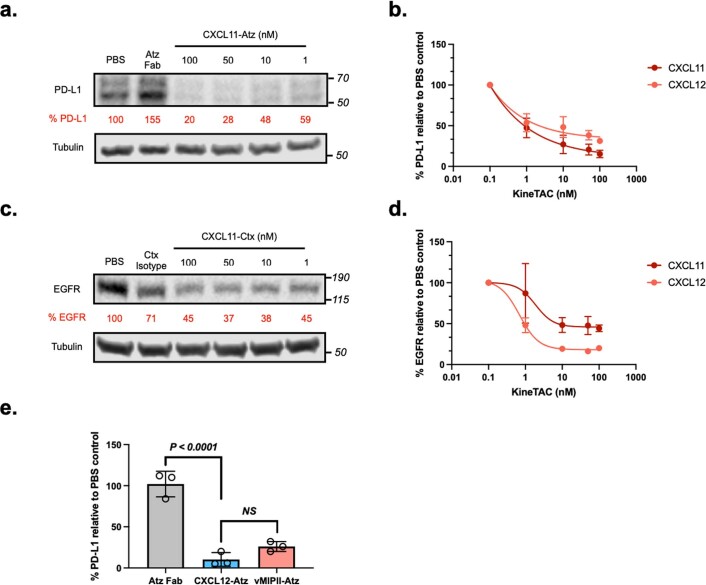
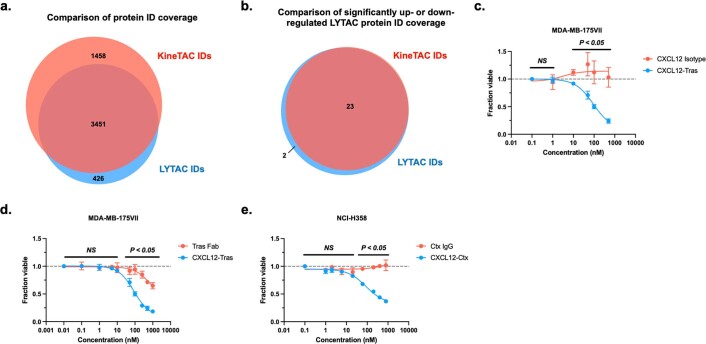
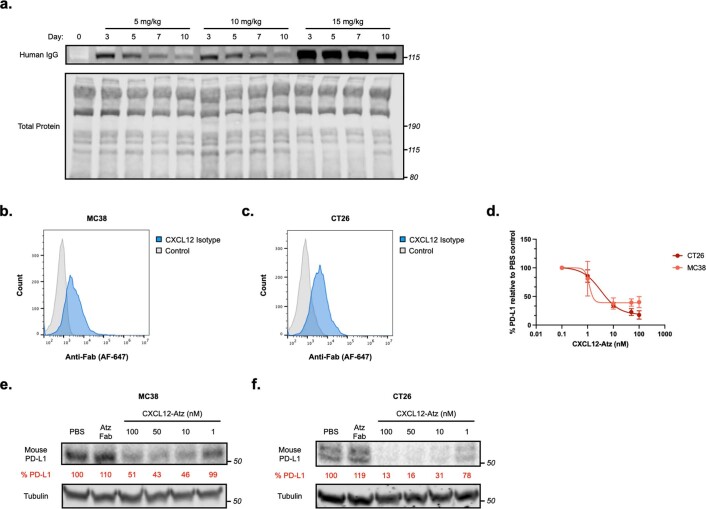
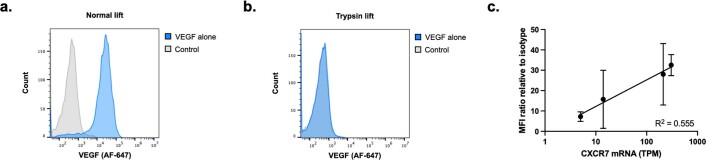
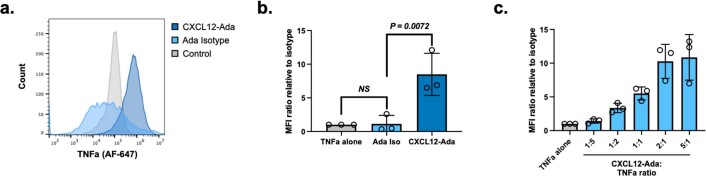
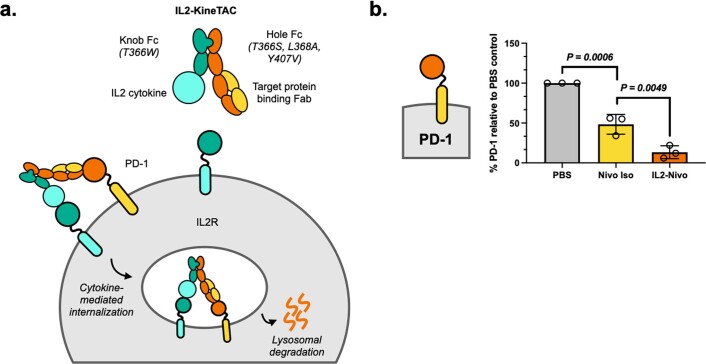
Targeted degradation of cell surface and extracellular proteins via lysosomal delivery is an important means to modulate extracellular biology. However, these approaches have limitations due to lack of modularity, ease of development, restricted tissue targeting and applicability to both cell surface and extracellular proteins. We describe a lysosomal degradation strategy, termed cytokine receptor-targeting chimeras (KineTACs), that addresses these limitations. KineTACs are fully genetically encoded bispecific antibodies consisting of a cytokine arm, which binds its cognate cytokine receptor, and a target-binding arm for the protein of interest. We show that KineTACs containing the cytokine CXCL12 can use the decoy recycling receptor, CXCR7, to target a variety of target proteins to the lysosome for degradation. Additional KineTACs were designed to harness other CXCR7-targeting cytokines, CXCL11 and vMIPII, and the interleukin-2 (IL-2) receptor-targeting cytokine IL-2. Thus, KineTACs represent a general, modular, selective and simple genetically encoded strategy for inducing lysosomal delivery of extracellular and cell surface targets with broad or tissue-specific distribution.
© 2022. The Author(s).
Conflict of interest statement
K.P., J.A.W. and J.A.G. and the Reagents of the University of California have filed a patent application related to this project. K.P. and J.A.G. are cofounders, shareholders and employees of EpiBiologics. J.A.W. is a cofounder, shareholder and board member of EpiBiologics. A.D.C. is a cofounder and shareholder of EpiBiologics. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
